 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeStructure-Based Drug Design via 3D Molecular Generative Pre-training and Sampling
Paper and Code
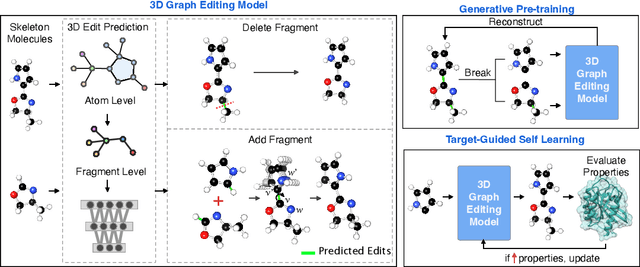
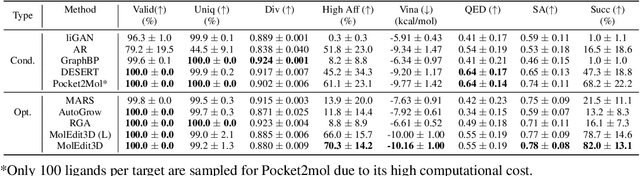
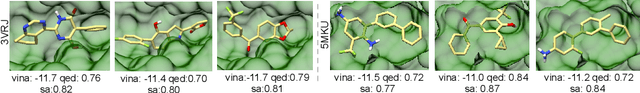
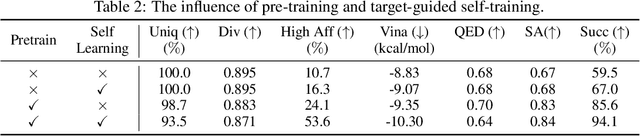
Structure-based drug design aims at generating high affinity ligands with prior knowledge of 3D target structures. Existing methods either use conditional generative model to learn the distribution of 3D ligands given target binding sites, or iteratively modify molecules to optimize a structure-based activity estimator. The former is highly constrained by data quantity and quality, which leaves optimization-based approaches more promising in practical scenario. However, existing optimization-based approaches choose to edit molecules in 2D space, and use molecular docking to estimate the activity using docking predicted 3D target-ligand complexes. The misalignment between the action space and the objective hinders the performance of these models, especially for those employ deep learning for acceleration. In this work, we propose MolEdit3D to combine 3D molecular generation with optimization frameworks. We develop a novel 3D graph editing model to generate molecules using fragments, and pre-train this model on abundant 3D ligands for learning target-independent properties. Then we employ a target-guided self-learning strategy to improve target-related properties using self-sampled molecules. MolEdit3D achieves state-of-the-art performance on majority of the evaluation metrics, and demonstrate strong capability of capturing both target-dependent and -independent properties.