 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMolChord: Structure-Sequence Alignment for Protein-Guided Drug Design
Oct 31, 2025Structure-based drug design (SBDD), which maps target proteins to candidate molecular ligands, is a fundamental task in drug discovery. Effectively aligning protein structural representations with molecular representations, and ensuring alignment between generated drugs and their pharmacological properties, remains a critical challenge. To address these challenges, we propose MolChord, which integrates two key techniques: (1) to align protein and molecule structures with their textual descriptions and sequential representations (e.g., FASTA for proteins and SMILES for molecules), we leverage NatureLM, an autoregressive model unifying text, small molecules, and proteins, as the molecule generator, alongside a diffusion-based structure encoder; and (2) to guide molecules toward desired properties, we curate a property-aware dataset by integrating preference data and refine the alignment process using Direct Preference Optimization (DPO). Experimental results on CrossDocked2020 demonstrate that our approach achieves state-of-the-art performance on key evaluation metrics, highlighting its potential as a practical tool for SBDD.
Trust Region Preference Approximation: A simple and stable reinforcement learning algorithm for LLM reasoning
Apr 06, 2025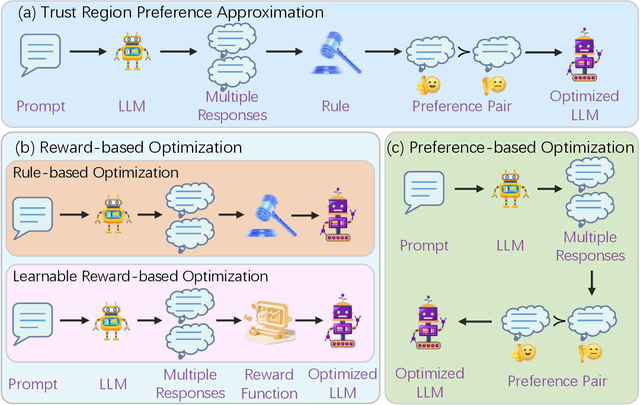
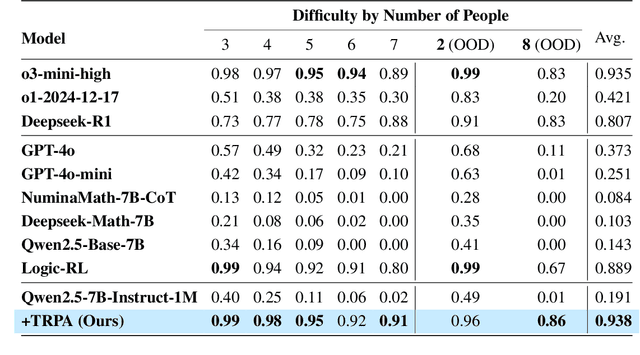
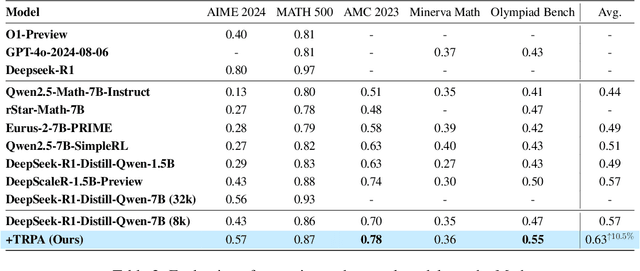
Recently, Large Language Models (LLMs) have rapidly evolved, approaching Artificial General Intelligence (AGI) while benefiting from large-scale reinforcement learning to enhance Human Alignment (HA) and Reasoning. Recent reward-based optimization algorithms, such as Proximal Policy Optimization (PPO) and Group Relative Policy Optimization (GRPO) have achieved significant performance on reasoning tasks, whereas preference-based optimization algorithms such as Direct Preference Optimization (DPO) significantly improve the performance of LLMs on human alignment. However, despite the strong performance of reward-based optimization methods in alignment tasks , they remain vulnerable to reward hacking. Furthermore, preference-based algorithms (such as Online DPO) haven't yet matched the performance of reward-based optimization algorithms (like PPO) on reasoning tasks, making their exploration in this specific area still a worthwhile pursuit. Motivated by these challenges, we propose the Trust Region Preference Approximation (TRPA) algorithm, which integrates rule-based optimization with preference-based optimization for reasoning tasks. As a preference-based algorithm, TRPA naturally eliminates the reward hacking issue. TRPA constructs preference levels using predefined rules, forms corresponding preference pairs, and leverages a novel optimization algorithm for RL training with a theoretical monotonic improvement guarantee. Experimental results demonstrate that TRPA not only achieves competitive performance on reasoning tasks but also exhibits robust stability. The code of this paper are released and updating on https://github.com/XueruiSu/Trust-Region-Preference-Approximation.git.
UniGenX: Unified Generation of Sequence and Structure with Autoregressive Diffusion
Mar 09, 2025Unified generation of sequence and structure for scientific data (e.g., materials, molecules, proteins) is a critical task. Existing approaches primarily rely on either autoregressive sequence models or diffusion models, each offering distinct advantages and facing notable limitations. Autoregressive models, such as GPT, Llama, and Phi-4, have demonstrated remarkable success in natural language generation and have been extended to multimodal tasks (e.g., image, video, and audio) using advanced encoders like VQ-VAE to represent complex modalities as discrete sequences. However, their direct application to scientific domains is challenging due to the high precision requirements and the diverse nature of scientific data. On the other hand, diffusion models excel at generating high-dimensional scientific data, such as protein, molecule, and material structures, with remarkable accuracy. Yet, their inability to effectively model sequences limits their potential as general-purpose multimodal foundation models. To address these challenges, we propose UniGenX, a unified framework that combines autoregressive next-token prediction with conditional diffusion models. This integration leverages the strengths of autoregressive models to ease the training of conditional diffusion models, while diffusion-based generative heads enhance the precision of autoregressive predictions. We validate the effectiveness of UniGenX on material and small molecule generation tasks, achieving a significant leap in state-of-the-art performance for material crystal structure prediction and establishing new state-of-the-art results for small molecule structure prediction, de novo design, and conditional generation. Notably, UniGenX demonstrates significant improvements, especially in handling long sequences for complex structures, showcasing its efficacy as a versatile tool for scientific data generation.
NatureLM: Deciphering the Language of Nature for Scientific Discovery
Feb 11, 2025



Foundation models have revolutionized natural language processing and artificial intelligence, significantly enhancing how machines comprehend and generate human languages. Inspired by the success of these foundation models, researchers have developed foundation models for individual scientific domains, including small molecules, materials, proteins, DNA, and RNA. However, these models are typically trained in isolation, lacking the ability to integrate across different scientific domains. Recognizing that entities within these domains can all be represented as sequences, which together form the "language of nature", we introduce Nature Language Model (briefly, NatureLM), a sequence-based science foundation model designed for scientific discovery. Pre-trained with data from multiple scientific domains, NatureLM offers a unified, versatile model that enables various applications including: (i) generating and optimizing small molecules, proteins, RNA, and materials using text instructions; (ii) cross-domain generation/design, such as protein-to-molecule and protein-to-RNA generation; and (iii) achieving state-of-the-art performance in tasks like SMILES-to-IUPAC translation and retrosynthesis on USPTO-50k. NatureLM offers a promising generalist approach for various scientific tasks, including drug discovery (hit generation/optimization, ADMET optimization, synthesis), novel material design, and the development of therapeutic proteins or nucleotides. We have developed NatureLM models in different sizes (1 billion, 8 billion, and 46.7 billion parameters) and observed a clear improvement in performance as the model size increases.
Exploiting Pre-trained Models for Drug Target Affinity Prediction with Nearest Neighbors
Jul 21, 2024



Drug-Target binding Affinity (DTA) prediction is essential for drug discovery. Despite the application of deep learning methods to DTA prediction, the achieved accuracy remain suboptimal. In this work, inspired by the recent success of retrieval methods, we propose $k$NN-DTA, a non-parametric embedding-based retrieval method adopted on a pre-trained DTA prediction model, which can extend the power of the DTA model with no or negligible cost. Different from existing methods, we introduce two neighbor aggregation ways from both embedding space and label space that are integrated into a unified framework. Specifically, we propose a \emph{label aggregation} with \emph{pair-wise retrieval} and a \emph{representation aggregation} with \emph{point-wise retrieval} of the nearest neighbors. This method executes in the inference phase and can efficiently boost the DTA prediction performance with no training cost. In addition, we propose an extension, Ada-$k$NN-DTA, an instance-wise and adaptive aggregation with lightweight learning. Results on four benchmark datasets show that $k$NN-DTA brings significant improvements, outperforming previous state-of-the-art (SOTA) results, e.g, on BindingDB IC$_{50}$ and $K_i$ testbeds, $k$NN-DTA obtains new records of RMSE $\bf{0.684}$ and $\bf{0.750}$. The extended Ada-$k$NN-DTA further improves the performance to be $\bf{0.675}$ and $\bf{0.735}$ RMSE. These results strongly prove the effectiveness of our method. Results in other settings and comprehensive studies/analyses also show the great potential of our $k$NN-DTA approach.
FABind: Fast and Accurate Protein-Ligand Binding
Oct 17, 2023Modeling the interaction between proteins and ligands and accurately predicting their binding structures is a critical yet challenging task in drug discovery. Recent advancements in deep learning have shown promise in addressing this challenge, with sampling-based and regression-based methods emerging as two prominent approaches. However, these methods have notable limitations. Sampling-based methods often suffer from low efficiency due to the need for generating multiple candidate structures for selection. On the other hand, regression-based methods offer fast predictions but may experience decreased accuracy. Additionally, the variation in protein sizes often requires external modules for selecting suitable binding pockets, further impacting efficiency. In this work, we propose $\mathbf{FABind}$, an end-to-end model that combines pocket prediction and docking to achieve accurate and fast protein-ligand binding. $\mathbf{FABind}$ incorporates a unique ligand-informed pocket prediction module, which is also leveraged for docking pose estimation. The model further enhances the docking process by incrementally integrating the predicted pocket to optimize protein-ligand binding, reducing discrepancies between training and inference. Through extensive experiments on benchmark datasets, our proposed $\mathbf{FABind}$ demonstrates strong advantages in terms of effectiveness and efficiency compared to existing methods. Our code is available at $\href{https://github.com/QizhiPei/FABind}{Github}$.
BioT5: Enriching Cross-modal Integration in Biology with Chemical Knowledge and Natural Language Associations
Oct 17, 2023



Recent advancements in biological research leverage the integration of molecules, proteins, and natural language to enhance drug discovery. However, current models exhibit several limitations, such as the generation of invalid molecular SMILES, underutilization of contextual information, and equal treatment of structured and unstructured knowledge. To address these issues, we propose $\mathbf{BioT5}$, a comprehensive pre-training framework that enriches cross-modal integration in biology with chemical knowledge and natural language associations. $\mathbf{BioT5}$ utilizes SELFIES for $100%$ robust molecular representations and extracts knowledge from the surrounding context of bio-entities in unstructured biological literature. Furthermore, $\mathbf{BioT5}$ distinguishes between structured and unstructured knowledge, leading to more effective utilization of information. After fine-tuning, BioT5 shows superior performance across a wide range of tasks, demonstrating its strong capability of capturing underlying relations and properties of bio-entities. Our code is available at $\href{https://github.com/QizhiPei/BioT5}{Github}$.
Retrosynthesis Prediction with Local Template Retrieval
Jun 07, 2023



Retrosynthesis, which predicts the reactants of a given target molecule, is an essential task for drug discovery. In recent years, the machine learing based retrosynthesis methods have achieved promising results. In this work, we introduce RetroKNN, a local reaction template retrieval method to further boost the performance of template-based systems with non-parametric retrieval. We first build an atom-template store and a bond-template store that contain the local templates in the training data, then retrieve from these templates with a k-nearest-neighbor (KNN) search during inference. The retrieved templates are combined with neural network predictions as the final output. Furthermore, we propose a lightweight adapter to adjust the weights when combing neural network and KNN predictions conditioned on the hidden representation and the retrieved templates. We conduct comprehensive experiments on two widely used benchmarks, the USPTO-50K and USPTO-MIT. Especially for the top-1 accuracy, we improved 7.1% on the USPTO-50K dataset and 12.0% on the USPTO-MIT dataset. These results demonstrate the effectiveness of our method.
MolXPT: Wrapping Molecules with Text for Generative Pre-training
May 26, 2023
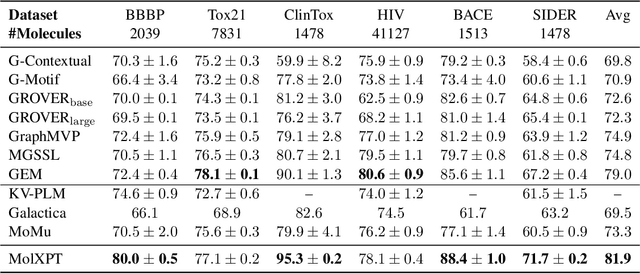
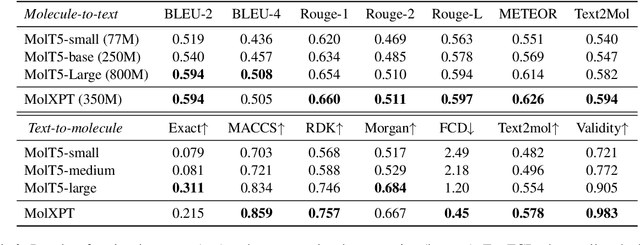

Generative pre-trained Transformer (GPT) has demonstrates its great success in natural language processing and related techniques have been adapted into molecular modeling. Considering that text is the most important record for scientific discovery, in this paper, we propose MolXPT, a unified language model of text and molecules pre-trained on SMILES (a sequence representation of molecules) wrapped by text. Briefly, we detect the molecule names in each sequence and replace them to the corresponding SMILES. In this way, the SMILES could leverage the information from surrounding text, and vice versa. The above wrapped sequences, text sequences from PubMed and SMILES sequences from PubChem are all fed into a language model for pre-training. Experimental results demonstrate that MolXPT outperforms strong baselines of molecular property prediction on MoleculeNet, performs comparably to the best model in text-molecule translation while using less than half of its parameters, and enables zero-shot molecular generation without finetuning.
What are the Desired Characteristics of Calibration Sets? Identifying Correlates on Long Form Scientific Summarization
May 12, 2023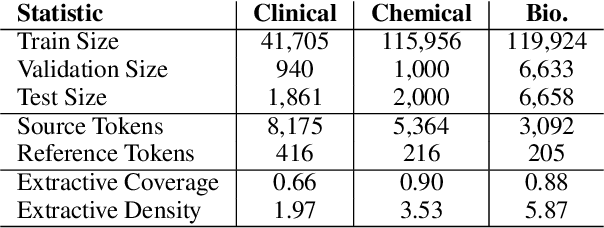
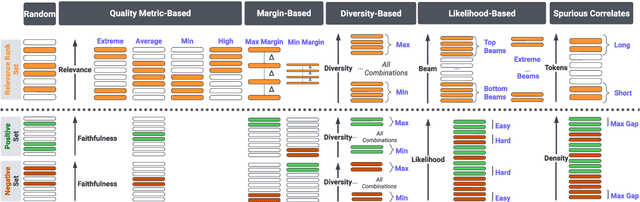
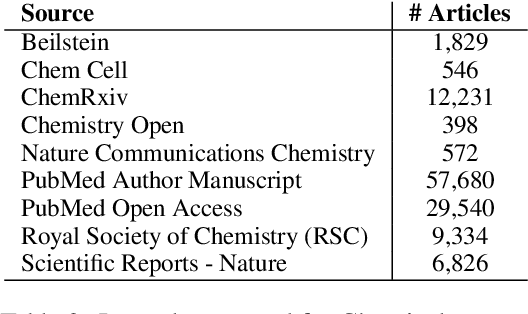
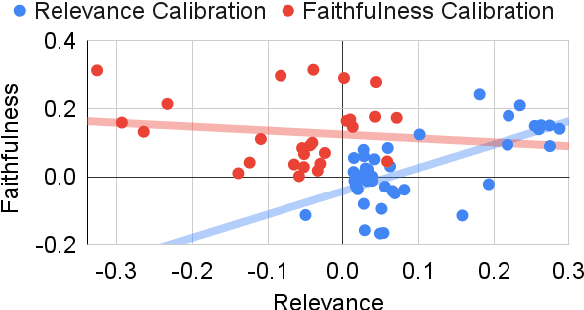
Summarization models often generate text that is poorly calibrated to quality metrics because they are trained to maximize the likelihood of a single reference (MLE). To address this, recent work has added a calibration step, which exposes a model to its own ranked outputs to improve relevance or, in a separate line of work, contrasts positive and negative sets to improve faithfulness. While effective, much of this work has focused on how to generate and optimize these sets. Less is known about why one setup is more effective than another. In this work, we uncover the underlying characteristics of effective sets. For each training instance, we form a large, diverse pool of candidates and systematically vary the subsets used for calibration fine-tuning. Each selection strategy targets distinct aspects of the sets, such as lexical diversity or the size of the gap between positive and negatives. On three diverse scientific long-form summarization datasets (spanning biomedical, clinical, and chemical domains), we find, among others, that faithfulness calibration is optimal when the negative sets are extractive and more likely to be generated, whereas for relevance calibration, the metric margin between candidates should be maximized and surprise--the disagreement between model and metric defined candidate rankings--minimized. Code to create, select, and optimize calibration sets is available at https://github.com/griff4692/calibrating-summaries