 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSeeing Isn't Knowing: Do VLMs Know When Not to Answer Spatial Questions (and Why)?
May 28, 2026Spatial reasoning is a fundamental capability for vision-language models (VLMs) deployed in real-world environments. However, visual observations are inherently limited representations of a 3D world: occlusion can render objects invisible, and perspective can make geometric properties misleading. Despite this, existing spatial reasoning benchmarks typically assume that observations are sufficient and reliable, focusing on whether models produce correct answers rather than whether they recognize when a question cannot be answered and what additional observations would be needed. In this work, we challenge this assumption by constructing a controlled evaluation framework, SpatialUncertain, and introducing two types of observation challenges: (1) occlusion, which hides target information, and (2) perspective ambiguity, which produces misleading visual cues. For each configuration, we design spatial questions that are answerable under clean observations but require abstention under the introduced challenges. We further evaluate whether models can identify which additional viewpoints would resolve perspective ambiguity. Our results across a diverse set of frontier open- and closed-source VLMs reveal two consistent failure modes. First, models are prone to overconfident answering, attempting to solve spatial reasoning tasks even when visual evidence is incomplete or misleading, with average accuracy around 30\% under occlusion and below 10\% under perspective ambiguity. Second, even when additional views are available, some models perform near random chance in identifying which would provide reliable evidence. Together, our findings call for moving beyond answer correctness toward evaluating whether models know when to abstain and how to seek reliable evidence.
Retrieval, Reward, and Training Protocols: What Matters in Training Search Agents?
May 27, 2026Search agents powered by large language models can autonomously decompose queries, retrieve information, and synthesize answers through multi-step reasoning. However, the rapid growth of training methods has outpaced controlled comparison: existing works differ in retrieval corpora, reward designs, and training protocols, making it unclear what actually drives improvements. We present a controlled empirical study that isolates three under-explored dimensions of search agent training. First, we identify a critical data-coverage issue in the widely used Wikipedia 2018 corpus and show that correcting it alone yields larger gains than the differences between training algorithms. Second, we systematically compare outcome-based and process-based reward methods across three base models, finding that the simplest outcome-based approach achieves competitive or superior performance in most settings, and that process-level credit assignment can over-correct agent behavior. Third, we analyze training data diversity, off-policy data utilization, and search budget scaling, distilling practical guidelines for training effective search agents. Our code is available at https://github.com/YiboZhao624/SearchAgentReview.
PhyMotion: Structured 3D Motion Reward for Physics-Grounded Human Video Generation
May 14, 2026Generating realistic human motion is a central yet unsolved challenge in video generation. While reinforcement learning (RL)-based post-training has driven recent gains in general video quality, extending it to human motion remains bottlenecked by a reward signal that cannot reliably score motion realism. Existing video rewards primarily rely on 2D perceptual signals, without explicitly modeling the 3D body state, contact, and dynamics underlying articulated human motion, and often assign high scores to videos with floating bodies or physically implausible movements. To address this, we propose PhyMotion, a structured, fine-grained motion reward that grounds recovered 3D human trajectories in a physics simulator and evaluates motion quality along multiple dimensions of physical feasibility. Concretely, we recover SMPL body meshes from generated videos, retarget them onto a humanoid in the MuJoCo physics simulator, and evaluate the resulting motion along three axes: kinematic plausibility, contact and balance consistency, and dynamic feasibility. Each component provides a continuous and interpretable signal tied to a specific aspect of motion quality, allowing the reward to capture which aspects of motion are physically correct or violated. Experiments show that PhyMotion achieves stronger correlation with human judgments than existing reward formulations. These gains carry over to RL-based post-training, where optimizing PhyMotion leads to larger and more consistent improvements than optimizing existing rewards, improving motion realism across both autoregressive and bidirectional video generators under both automatic metrics and blind human evaluation (+68 Elo gain). Ablations show that the three axes provide complementary supervision signals, while the reward preserves overall video generation quality with only modest training overhead.
Intern-S1-Pro: Scientific Multimodal Foundation Model at Trillion Scale
Mar 26, 2026We introduce Intern-S1-Pro, the first one-trillion-parameter scientific multimodal foundation model. Scaling to this unprecedented size, the model delivers a comprehensive enhancement across both general and scientific domains. Beyond stronger reasoning and image-text understanding capabilities, its intelligence is augmented with advanced agent capabilities. Simultaneously, its scientific expertise has been vastly expanded to master over 100 specialized tasks across critical science fields, including chemistry, materials, life sciences, and earth sciences. Achieving this massive scale is made possible by the robust infrastructure support of XTuner and LMDeploy, which facilitates highly efficient Reinforcement Learning (RL) training at the 1-trillion parameter level while ensuring strict precision consistency between training and inference. By seamlessly integrating these advancements, Intern-S1-Pro further fortifies the fusion of general and specialized intelligence, working as a Specializable Generalist, demonstrating its position in the top tier of open-source models for general capabilities, while outperforming proprietary models in the depth of specialized scientific tasks.
OS-Themis: A Scalable Critic Framework for Generalist GUI Rewards
Mar 19, 2026Reinforcement Learning (RL) has the potential to improve the robustness of GUI agents in stochastic environments, yet training is highly sensitive to the quality of the reward function. Existing reward approaches struggle to achieve both scalability and performance. To address this, we propose OS-Themis, a scalable and accurate multi-agent critic framework. Unlike a single judge, OS-Themis decomposes trajectories into verifiable milestones to isolate critical evidence for decision making and employs a review mechanism to strictly audit the evidence chain before making the final verdict. To facilitate evaluation, we further introduce OmniGUIRewardBench (OGRBench), a holistic cross-platform benchmark for GUI outcome rewards, where all evaluated models achieve their best performance under OS-Themis. Extensive experiments on AndroidWorld show that OS-Themis yields a 10.3% improvement when used to support online RL training, and a 6.9% gain when used for trajectory validation and filtering in the self-training loop, highlighting its potential to drive agent evolution.
V-Co: A Closer Look at Visual Representation Alignment via Co-Denoising
Mar 17, 2026Pixel-space diffusion has recently re-emerged as a strong alternative to latent diffusion, enabling high-quality generation without pretrained autoencoders. However, standard pixel-space diffusion models receive relatively weak semantic supervision and are not explicitly designed to capture high-level visual structure. Recent representation-alignment methods (e.g., REPA) suggest that pretrained visual features can substantially improve diffusion training, and visual co-denoising has emerged as a promising direction for incorporating such features into the generative process. However, existing co-denoising approaches often entangle multiple design choices, making it unclear which design choices are truly essential. Therefore, we present V-Co, a systematic study of visual co-denoising in a unified JiT-based framework. This controlled setting allows us to isolate the ingredients that make visual co-denoising effective. Our study reveals four key ingredients for effective visual co-denoising. First, preserving feature-specific computation while enabling flexible cross-stream interaction motivates a fully dual-stream architecture. Second, effective classifier-free guidance (CFG) requires a structurally defined unconditional prediction. Third, stronger semantic supervision is best provided by a perceptual-drifting hybrid loss. Fourth, stable co-denoising further requires proper cross-stream calibration, which we realize through RMS-based feature rescaling. Together, these findings yield a simple recipe for visual co-denoising. Experiments on ImageNet-256 show that, at comparable model sizes, V-Co outperforms the underlying pixel-space diffusion baseline and strong prior pixel-diffusion methods while using fewer training epochs, offering practical guidance for future representation-aligned generative models.
AnchorWeave: World-Consistent Video Generation with Retrieved Local Spatial Memories
Feb 16, 2026Maintaining spatial world consistency over long horizons remains a central challenge for camera-controllable video generation. Existing memory-based approaches often condition generation on globally reconstructed 3D scenes by rendering anchor videos from the reconstructed geometry in the history. However, reconstructing a global 3D scene from multiple views inevitably introduces cross-view misalignment, as pose and depth estimation errors cause the same surfaces to be reconstructed at slightly different 3D locations across views. When fused, these inconsistencies accumulate into noisy geometry that contaminates the conditioning signals and degrades generation quality. We introduce AnchorWeave, a memory-augmented video generation framework that replaces a single misaligned global memory with multiple clean local geometric memories and learns to reconcile their cross-view inconsistencies. To this end, AnchorWeave performs coverage-driven local memory retrieval aligned with the target trajectory and integrates the selected local memories through a multi-anchor weaving controller during generation. Extensive experiments demonstrate that AnchorWeave significantly improves long-term scene consistency while maintaining strong visual quality, with ablation and analysis studies further validating the effectiveness of local geometric conditioning, multi-anchor control, and coverage-driven retrieval.
When and How Much to Imagine: Adaptive Test-Time Scaling with World Models for Visual Spatial Reasoning
Feb 09, 2026Despite rapid progress in Multimodal Large Language Models (MLLMs), visual spatial reasoning remains unreliable when correct answers depend on how a scene would appear under unseen or alternative viewpoints. Recent work addresses this by augmenting reasoning with world models for visual imagination, but questions such as when imagination is actually necessary, how much of it is beneficial, and when it becomes harmful, remain poorly understood. In practice, indiscriminate imagination can increase computation and even degrade performance by introducing misleading evidence. In this work, we present an in-depth analysis of test-time visual imagination as a controllable resource for spatial reasoning. We study when static visual evidence is sufficient, when imagination improves reasoning, and how excessive or unnecessary imagination affects accuracy and efficiency. To support this analysis, we introduce AVIC, an adaptive test-time framework with world models that explicitly reasons about the sufficiency of current visual evidence before selectively invoking and scaling visual imagination. Across spatial reasoning benchmarks (SAT, MMSI) and an embodied navigation benchmark (R2R), our results reveal clear scenarios where imagination is critical, marginal, or detrimental, and show that selective control can match or outperform fixed imagination strategies with substantially fewer world-model calls and language tokens. Overall, our findings highlight the importance of analyzing and controlling test-time imagination for efficient and reliable spatial reasoning.
OS-Symphony: A Holistic Framework for Robust and Generalist Computer-Using Agent
Jan 12, 2026While Vision-Language Models (VLMs) have significantly advanced Computer-Using Agents (CUAs), current frameworks struggle with robustness in long-horizon workflows and generalization in novel domains. These limitations stem from a lack of granular control over historical visual context curation and the absence of visual-aware tutorial retrieval. To bridge these gaps, we introduce OS-Symphony, a holistic framework that comprises an Orchestrator coordinating two key innovations for robust automation: (1) a Reflection-Memory Agent that utilizes milestone-driven long-term memory to enable trajectory-level self-correction, effectively mitigating visual context loss in long-horizon tasks; (2) Versatile Tool Agents featuring a Multimodal Searcher that adopts a SeeAct paradigm to navigate a browser-based sandbox to synthesize live, visually aligned tutorials, thereby resolving fidelity issues in unseen scenarios. Experimental results demonstrate that OS-Symphony delivers substantial performance gains across varying model scales, establishing new state-of-the-art results on three online benchmarks, notably achieving 65.84% on OSWorld.
OS-Oracle: A Comprehensive Framework for Cross-Platform GUI Critic Models
Dec 18, 2025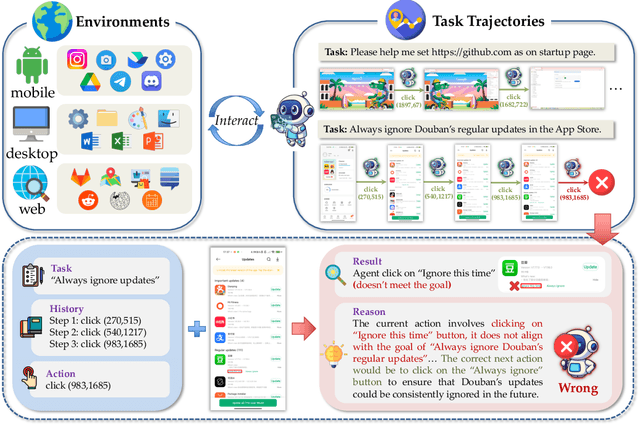
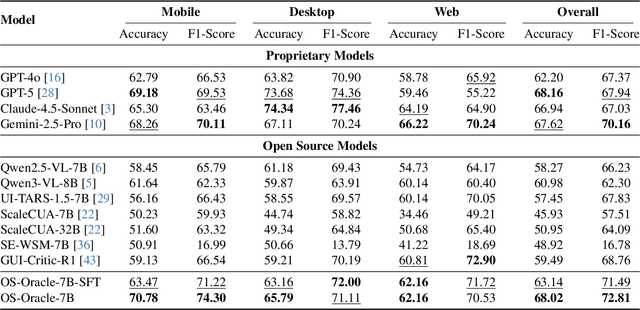
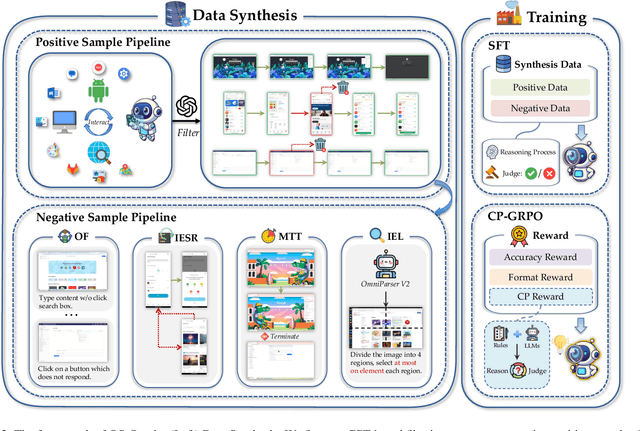
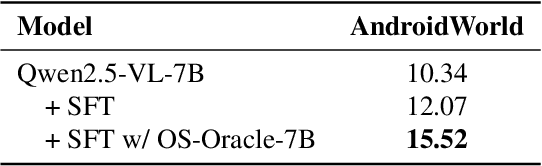
With VLM-powered computer-using agents (CUAs) becoming increasingly capable at graphical user interface (GUI) navigation and manipulation, reliable step-level decision-making has emerged as a key bottleneck for real-world deployment. In long-horizon workflows, errors accumulate quickly and irreversible actions can cause unintended consequences, motivating critic models that assess each action before execution. While critic models offer a promising solution, their effectiveness is hindered by the lack of diverse, high-quality GUI feedback data and public critic benchmarks for step-level evaluation in computer use. To bridge these gaps, we introduce OS-Oracle that makes three core contributions: (1) a scalable data pipeline for synthesizing cross-platform GUI critic data; (2) a two-stage training paradigm combining supervised fine-tuning (SFT) and consistency-preserving group relative policy optimization (CP-GRPO); (3) OS-Critic Bench, a holistic benchmark for evaluating critic model performance across Mobile, Web, and Desktop platforms. Leveraging this framework, we curate a high-quality dataset containing 310k critic samples. The resulting critic model, OS-Oracle-7B, achieves state-of-the-art performance among open-source VLMs on OS-Critic Bench, and surpasses proprietary models on the mobile domain. Furthermore, when serving as a pre-critic, OS-Oracle-7B improves the performance of native GUI agents such as UI-TARS-1.5-7B in OSWorld and AndroidWorld environments. The code is open-sourced at https://github.com/numbmelon/OS-Oracle.