 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeChallenging reaction prediction models to generalize to novel chemistry
Jan 11, 2025Deep learning models for anticipating the products of organic reactions have found many use cases, including validating retrosynthetic pathways and constraining synthesis-based molecular design tools. Despite compelling performance on popular benchmark tasks, strange and erroneous predictions sometimes ensue when using these models in practice. The core issue is that common benchmarks test models in an in-distribution setting, whereas many real-world uses for these models are in out-of-distribution settings and require a greater degree of extrapolation. To better understand how current reaction predictors work in out-of-distribution domains, we report a series of more challenging evaluations of a prototypical SMILES-based deep learning model. First, we illustrate how performance on randomly sampled datasets is overly optimistic compared to performance when generalizing to new patents or new authors. Second, we conduct time splits that evaluate how models perform when tested on reactions published in years after those in their training set, mimicking real-world deployment. Finally, we consider extrapolation across reaction classes to reflect what would be required for the discovery of novel reaction types. This panel of tasks can reveal the capabilities and limitations of today's reaction predictors, acting as a crucial first step in the development of tomorrow's next-generation models capable of reaction discovery.
Pareto Optimization to Accelerate Multi-Objective Virtual Screening
Oct 16, 2023The discovery of therapeutic molecules is fundamentally a multi-objective optimization problem. One formulation of the problem is to identify molecules that simultaneously exhibit strong binding affinity for a target protein, minimal off-target interactions, and suitable pharmacokinetic properties. Inspired by prior work that uses active learning to accelerate the identification of strong binders, we implement multi-objective Bayesian optimization to reduce the computational cost of multi-property virtual screening and apply it to the identification of ligands predicted to be selective based on docking scores to on- and off-targets. We demonstrate the superiority of Pareto optimization over scalarization across three case studies. Further, we use the developed optimization tool to search a virtual library of over 4M molecules for those predicted to be selective dual inhibitors of EGFR and IGF1R, acquiring 100% of the molecules that form the library's Pareto front after exploring only 8% of the library. This workflow and associated open source software can reduce the screening burden of molecular design projects and is complementary to research aiming to improve the accuracy of binding predictions and other molecular properties.
Self-focusing virtual screening with active design space pruning
May 03, 2022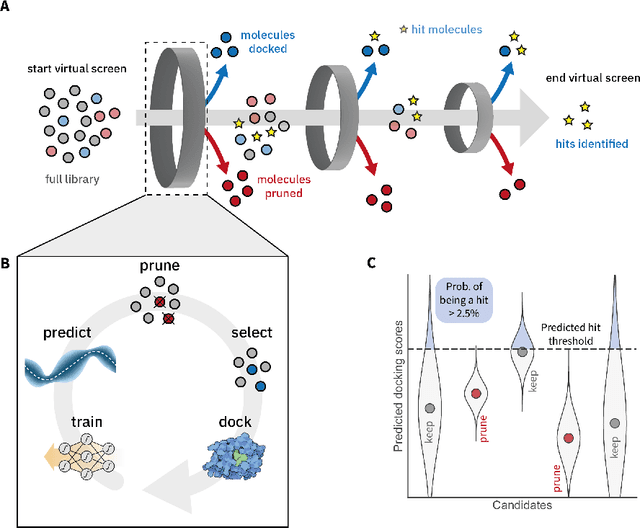
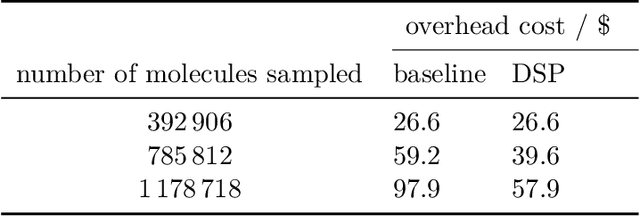
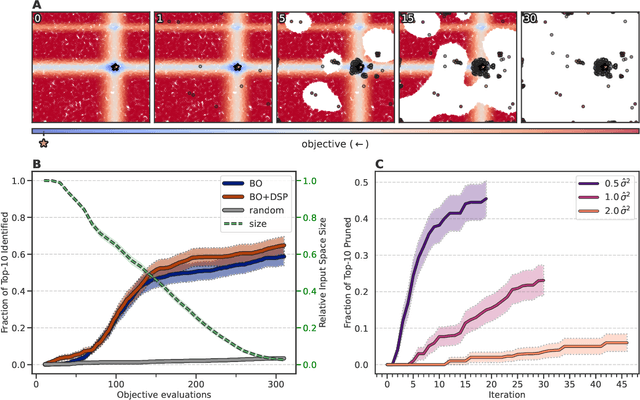
High-throughput virtual screening is an indispensable technique utilized in the discovery of small molecules. In cases where the library of molecules is exceedingly large, the cost of an exhaustive virtual screen may be prohibitive. Model-guided optimization has been employed to lower these costs through dramatic increases in sample efficiency compared to random selection. However, these techniques introduce new costs to the workflow through the surrogate model training and inference steps. In this study, we propose an extension to the framework of model-guided optimization that mitigates inferences costs using a technique we refer to as design space pruning (DSP), which irreversibly removes poor-performing candidates from consideration. We study the application of DSP to a variety of optimization tasks and observe significant reductions in overhead costs while exhibiting similar performance to the baseline optimization. DSP represents an attractive extension of model-guided optimization that can limit overhead costs in optimization settings where these costs are non-negligible relative to objective costs, such as docking.
Accelerating high-throughput virtual screening through molecular pool-based active learning
Dec 13, 2020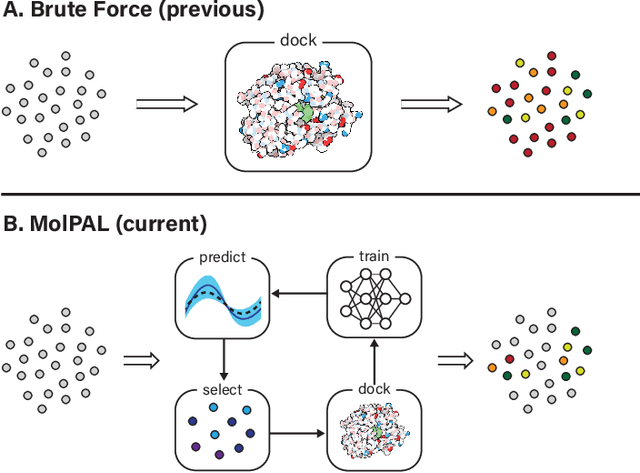


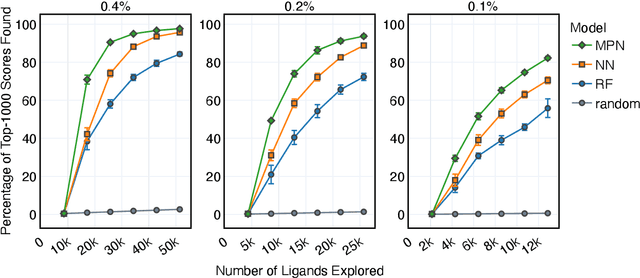
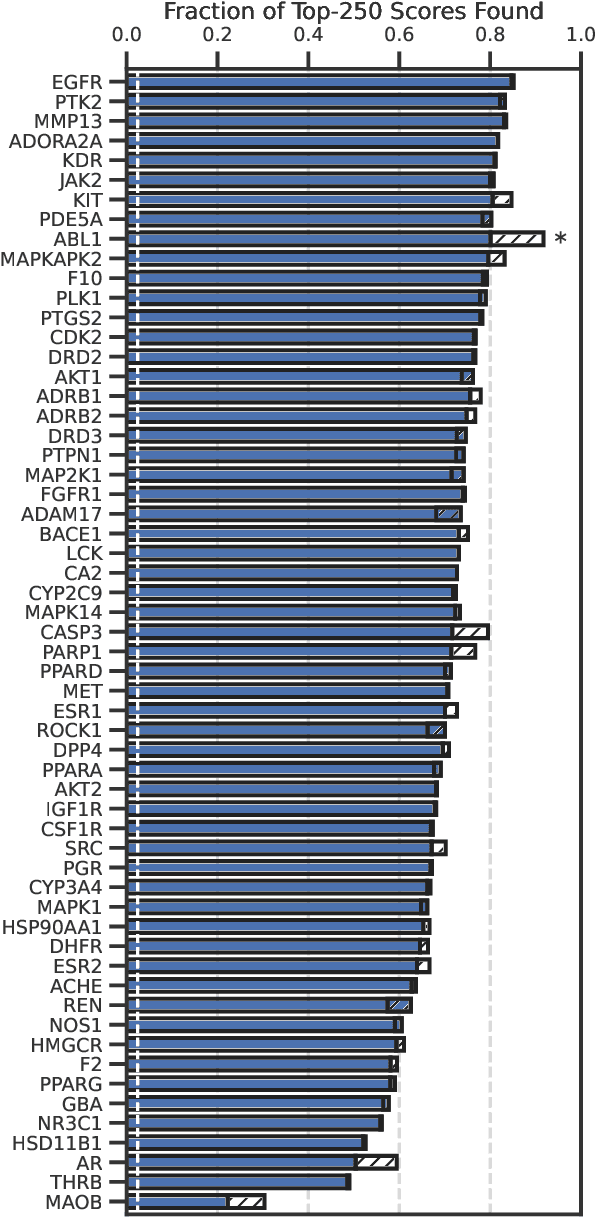
Structure-based virtual screening is an important tool in early stage drug discovery that scores the interactions between a target protein and candidate ligands. As virtual libraries continue to grow (in excess of $10^8$ molecules), so too do the resources necessary to conduct exhaustive virtual screening campaigns on these libraries. However, Bayesian optimization techniques can aid in their exploration: a surrogate structure-property relationship model trained on the predicted affinities of a subset of the library can be applied to the remaining library members, allowing the least promising compounds to be excluded from evaluation. In this study, we assess various surrogate model architectures, acquisition functions, and acquisition batch sizes as applied to several protein-ligand docking datasets and observe significant reductions in computational costs, even when using a greedy acquisition strategy; for example, 87.9% of the top-50000 ligands can be found after testing only 2.4% of a 100M member library. Such model-guided searches mitigate the increasing computational costs of screening increasingly large virtual libraries and can accelerate high-throughput virtual screening campaigns with applications beyond docking.