 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeChallenging reaction prediction models to generalize to novel chemistry
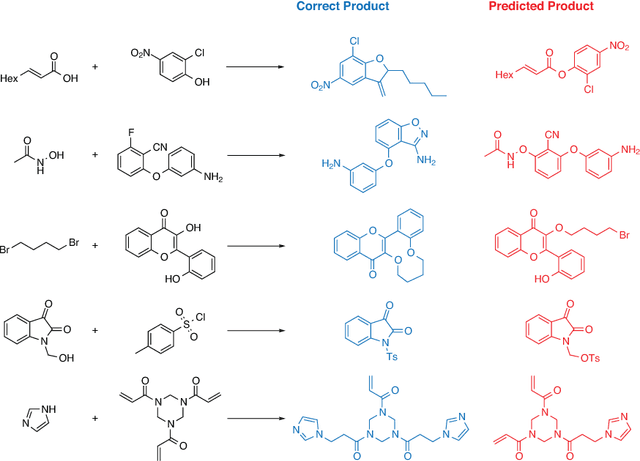
Jan 11, 2025Deep learning models for anticipating the products of organic reactions have found many use cases, including validating retrosynthetic pathways and constraining synthesis-based molecular design tools. Despite compelling performance on popular benchmark tasks, strange and erroneous predictions sometimes ensue when using these models in practice. The core issue is that common benchmarks test models in an in-distribution setting, whereas many real-world uses for these models are in out-of-distribution settings and require a greater degree of extrapolation. To better understand how current reaction predictors work in out-of-distribution domains, we report a series of more challenging evaluations of a prototypical SMILES-based deep learning model. First, we illustrate how performance on randomly sampled datasets is overly optimistic compared to performance when generalizing to new patents or new authors. Second, we conduct time splits that evaluate how models perform when tested on reactions published in years after those in their training set, mimicking real-world deployment. Finally, we consider extrapolation across reaction classes to reflect what would be required for the discovery of novel reaction types. This panel of tasks can reveal the capabilities and limitations of today's reaction predictors, acting as a crucial first step in the development of tomorrow's next-generation models capable of reaction discovery.
Barking up the right tree: an approach to search over molecule synthesis DAGs
Dec 21, 2020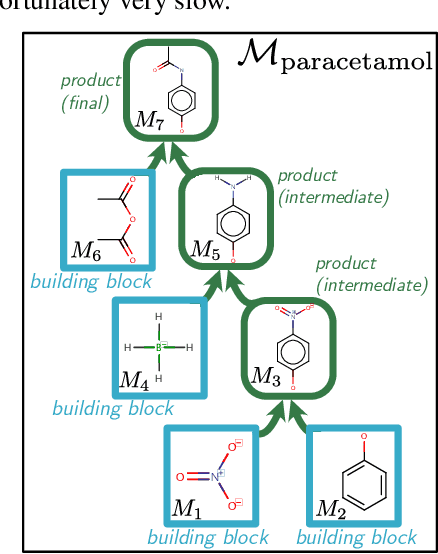
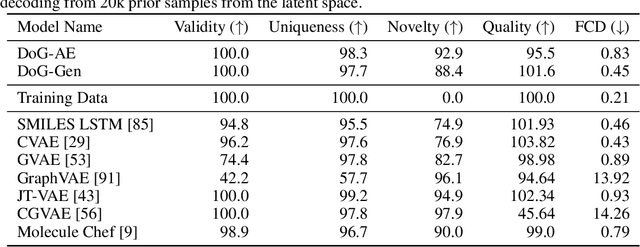
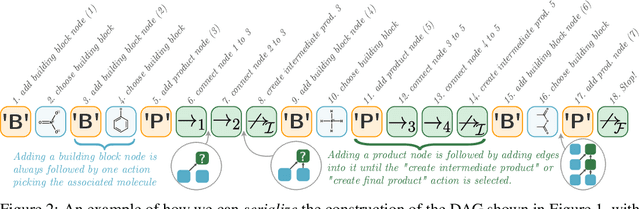
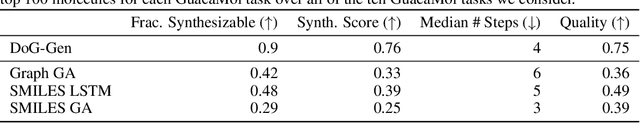
When designing new molecules with particular properties, it is not only important what to make but crucially how to make it. These instructions form a synthesis directed acyclic graph (DAG), describing how a large vocabulary of simple building blocks can be recursively combined through chemical reactions to create more complicated molecules of interest. In contrast, many current deep generative models for molecules ignore synthesizability. We therefore propose a deep generative model that better represents the real world process, by directly outputting molecule synthesis DAGs. We argue that this provides sensible inductive biases, ensuring that our model searches over the same chemical space that chemists would also have access to, as well as interpretability. We show that our approach is able to model chemical space well, producing a wide range of diverse molecules, and allows for unconstrained optimization of an inherently constrained problem: maximize certain chemical properties such that discovered molecules are synthesizable.
RetroGNN: Approximating Retrosynthesis by Graph Neural Networks for De Novo Drug Design
Nov 25, 2020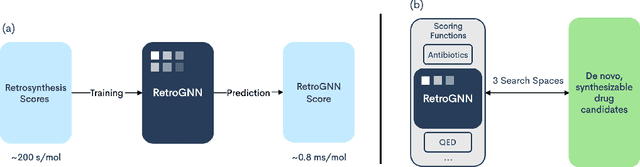
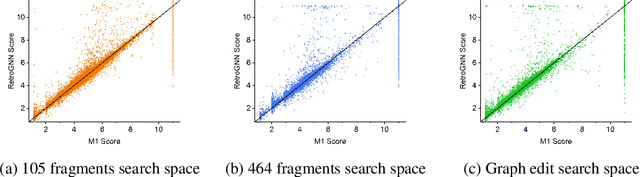
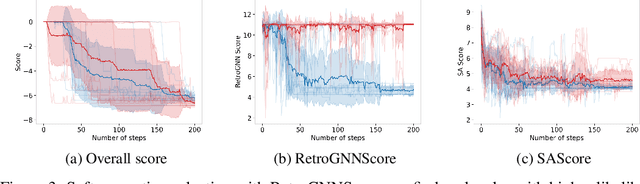
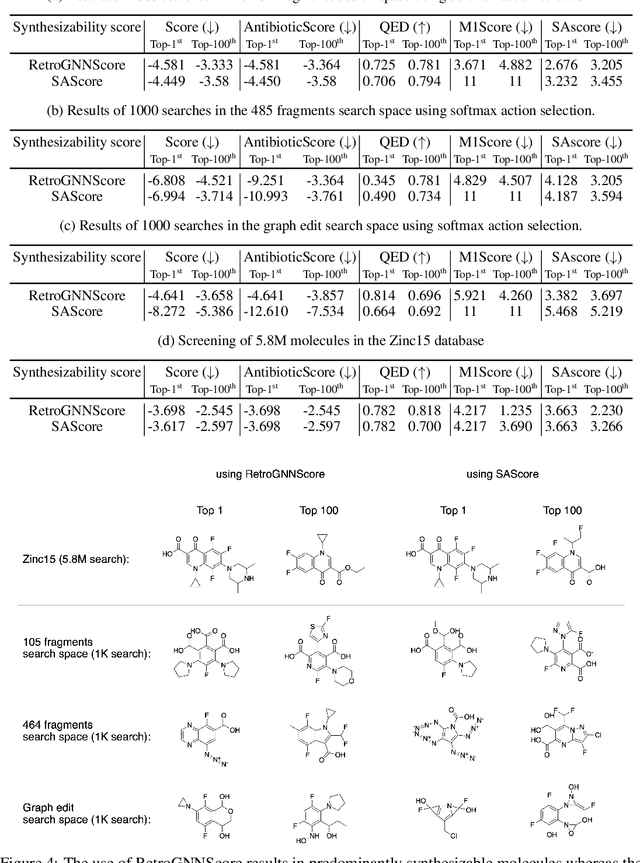
De novo molecule generation often results in chemically unfeasible molecules. A natural idea to mitigate this problem is to bias the search process towards more easily synthesizable molecules using a proxy for synthetic accessibility. However, using currently available proxies still results in highly unrealistic compounds. We investigate the feasibility of training deep graph neural networks to approximate the outputs of a retrosynthesis planning software, and their use to bias the search process. We evaluate our method on a benchmark involving searching for drug-like molecules with antibiotic properties. Compared to enumerating over five million existing molecules from the ZINC database, our approach finds molecules predicted to be more likely to be antibiotics while maintaining good drug-like properties and being easily synthesizable. Importantly, our deep neural network can successfully filter out hard to synthesize molecules while achieving a $10^5$ times speed-up over using the retrosynthesis planning software.
World Programs for Model-Based Learning and Planning in Compositional State and Action Spaces
Dec 30, 2019


Some of the most important tasks take place in environments which lack cheap and perfect simulators, thus hampering the application of model-free reinforcement learning (RL). While model-based RL aims to learn a dynamics model, in a more general case the learner does not know a priori what the action space is. Here we propose a formalism where the learner induces a world program by learning a dynamics model and the actions in graph-based compositional environments by observing state-state transition examples. Then, the learner can perform RL with the world program as the simulator for complex planning tasks. We highlight a recent application, and propose a challenge for the community to assess world program-based planning.
* Accepted at the Generative Modeling and Model-Based Reasoning for Robotics and AI workshop at ICML 2019. Presented on June 14th 2019. See https://sites.google.com/view/mbrl-icml2019
A Model to Search for Synthesizable Molecules
Jun 12, 2019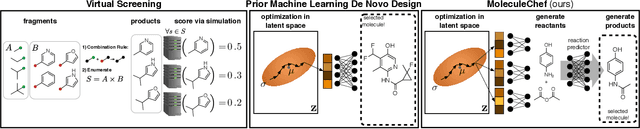
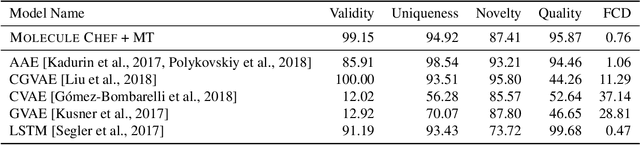
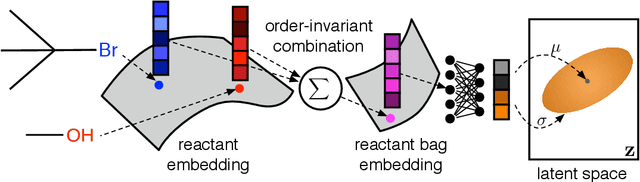
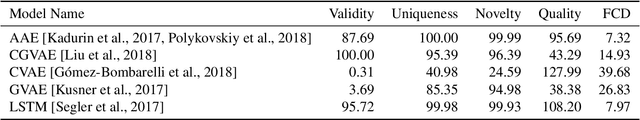
Deep generative models are able to suggest new organic molecules by generating strings, trees, and graphs representing their structure. While such models allow one to generate molecules with desirable properties, they give no guarantees that the molecules can actually be synthesized in practice. We propose a new molecule generation model, mirroring a more realistic real-world process, where (a) reactants are selected, and (b) combined to form more complex molecules. More specifically, our generative model proposes a bag of initial reactants (selected from a pool of commercially-available molecules) and uses a reaction model to predict how they react together to generate new molecules. We first show that the model can generate diverse, valid and unique molecules due to the useful inductive biases of modeling reactions. Furthermore, our model allows chemists to interrogate not only the properties of the generated molecules but also the feasibility of the synthesis routes. We conclude by using our model to solve retrosynthesis problems, predicting a set of reactants that can produce a target product.
GuacaMol: Benchmarking Models for De Novo Molecular Design
Nov 22, 2018
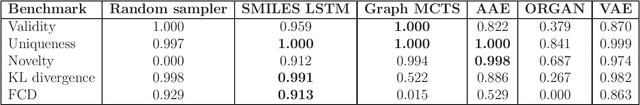
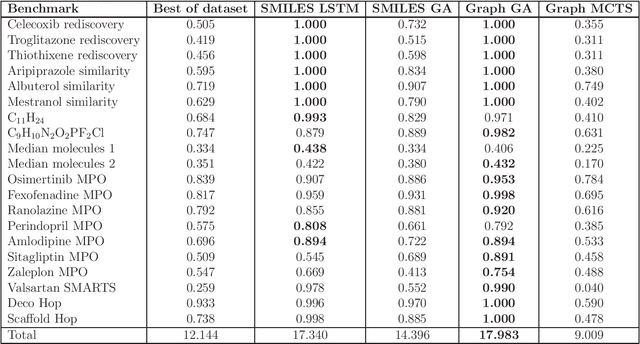
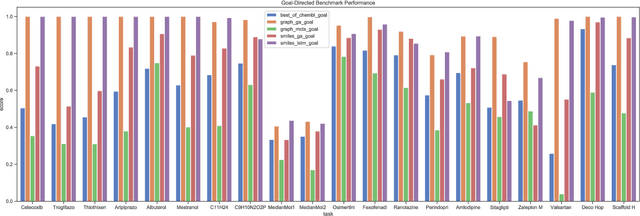
De novo design seeks to generate molecules with required property profiles by virtual design-make-test cycles. With the emergence of deep learning and neural generative models in many application areas, models for molecular design based on neural networks appeared recently and show promising results. However, the new models have not been profiled on consistent tasks, and comparative studies to well-established algorithms have only seldom been performed. To standardize the assessment of both classical and neural models for de novo molecular design, we propose an evaluation framework, GuacaMol, based on a suite of standardized benchmarks. The benchmark tasks encompass measuring the fidelity of the models to reproduce the property distribution of the training sets, the ability to generate novel molecules, the exploration and exploitation of chemical space, and a variety of single and multi-objective optimization tasks. The benchmarking framework is available as an open-source Python package.
Predicting Electron Paths
May 23, 2018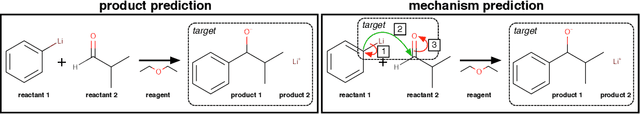
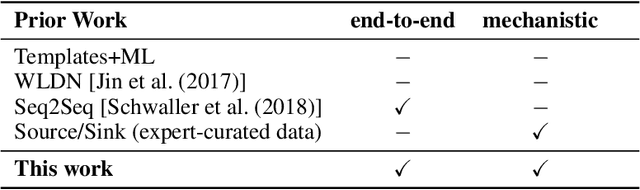
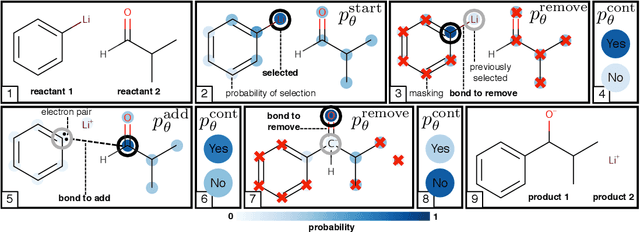
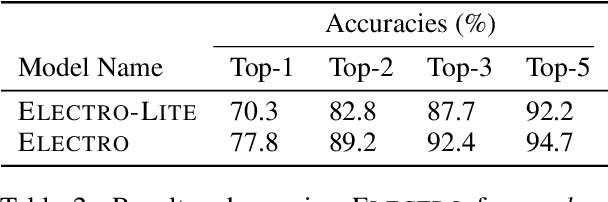
Chemical reactions can be described as the stepwise redistribution of electrons in molecules. As such, reactions are often depicted using "arrow-pushing" diagrams which show this movement as a sequence of arrows. We propose an electron path prediction model (ELECTRO) to learn these sequences directly from raw reaction data. Instead of predicting product molecules directly from reactant molecules in one shot, learning a model of electron movement has the benefits of (a) being easy for chemists to interpret, (b) incorporating constraints of chemistry, such as balanced atom counts before and after the reaction, and (c) naturally encoding the sparsity of chemical reactions, which usually involve changes in only a small number of atoms in the reactants. We design a method to extract approximate reaction paths from any dataset of atom-mapped reaction SMILES strings. Our model achieves state-of-the-art results on a subset of the UPSTO reaction dataset. Furthermore, we show that our model recovers a basic knowledge of chemistry without being explicitly trained to do so.
Learning to Plan Chemical Syntheses
Aug 14, 2017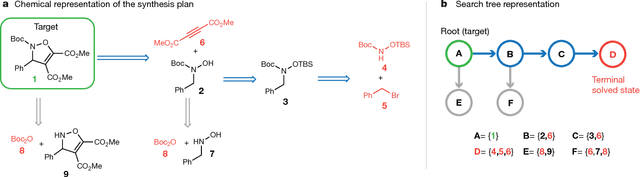
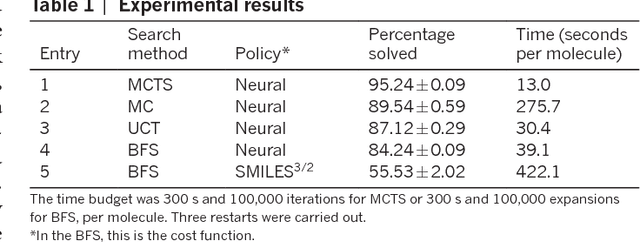
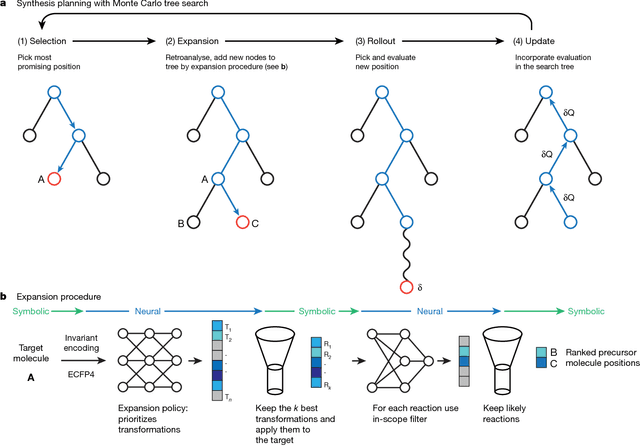
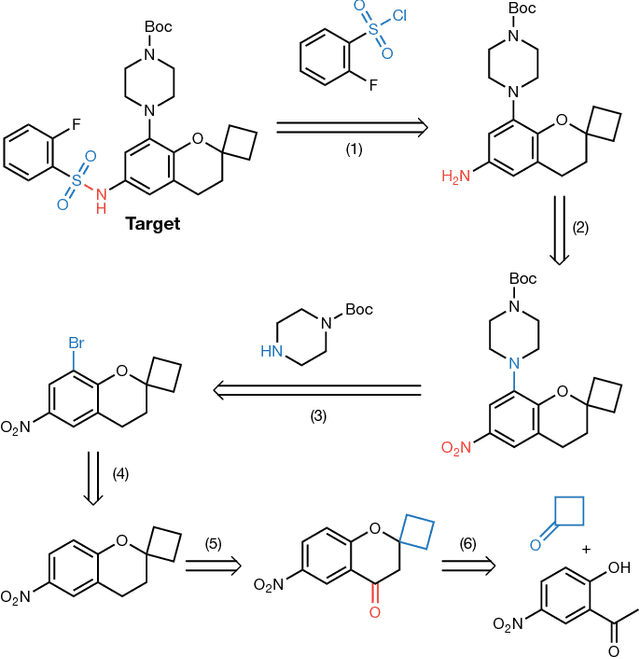
From medicines to materials, small organic molecules are indispensable for human well-being. To plan their syntheses, chemists employ a problem solving technique called retrosynthesis. In retrosynthesis, target molecules are recursively transformed into increasingly simpler precursor compounds until a set of readily available starting materials is obtained. Computer-aided retrosynthesis would be a highly valuable tool, however, past approaches were slow and provided results of unsatisfactory quality. Here, we employ Monte Carlo Tree Search (MCTS) to efficiently discover retrosynthetic routes. MCTS was combined with an expansion policy network that guides the search, and an "in-scope" filter network to pre-select the most promising retrosynthetic steps. These deep neural networks were trained on 12 million reactions, which represents essentially all reactions ever published in organic chemistry. Our system solves almost twice as many molecules and is 30 times faster in comparison to the traditional search method based on extracted rules and hand-coded heuristics. Finally after a 60 year history of computer-aided synthesis planning, chemists can no longer distinguish between routes generated by a computer system and real routes taken from the scientific literature. We anticipate that our method will accelerate drug and materials discovery by assisting chemists to plan better syntheses faster, and by enabling fully automated robot synthesis.
Generating Focussed Molecule Libraries for Drug Discovery with Recurrent Neural Networks
Jan 05, 2017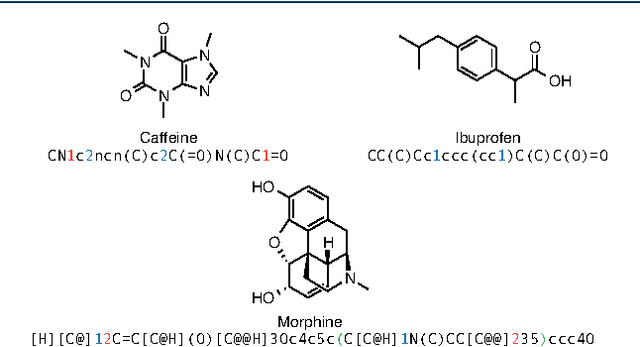

In de novo drug design, computational strategies are used to generate novel molecules with good affinity to the desired biological target. In this work, we show that recurrent neural networks can be trained as generative models for molecular structures, similar to statistical language models in natural language processing. We demonstrate that the properties of the generated molecules correlate very well with the properties of the molecules used to train the model. In order to enrich libraries with molecules active towards a given biological target, we propose to fine-tune the model with small sets of molecules, which are known to be active against that target. Against Staphylococcus aureus, the model reproduced 14% of 6051 hold-out test molecules that medicinal chemists designed, whereas against Plasmodium falciparum (Malaria) it reproduced 28% of 1240 test molecules. When coupled with a scoring function, our model can perform the complete de novo drug design cycle to generate large sets of novel molecules for drug discovery.
Modelling Chemical Reasoning to Predict Reactions
Aug 25, 2016


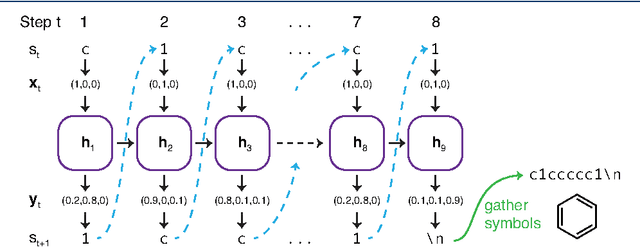
The ability to reason beyond established knowledge allows Organic Chemists to solve synthetic problems and to invent novel transformations. Here, we propose a model which mimics chemical reasoning and formalises reaction prediction as finding missing links in a knowledge graph. We have constructed a knowledge graph containing 14.4 million molecules and 8.2 million binary reactions, which represents the bulk of all chemical reactions ever published in the scientific literature. Our model outperforms a rule-based expert system in the reaction prediction task for 180,000 randomly selected binary reactions. We show that our data-driven model generalises even beyond known reaction types, and is thus capable of effectively (re-) discovering novel transformations (even including transition-metal catalysed reactions). Our model enables computers to infer hypotheses about reactivity and reactions by only considering the intrinsic local structure of the graph, and because each single reaction prediction is typically achieved in a sub-second time frame, our model can be used as a high-throughput generator of reaction hypotheses for reaction discovery.
* 17 pages, 8 figures