 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMolecular representation learning with language models and domain-relevant auxiliary tasks
Nov 26, 2020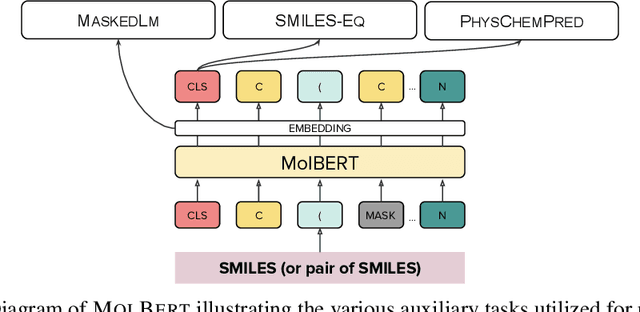

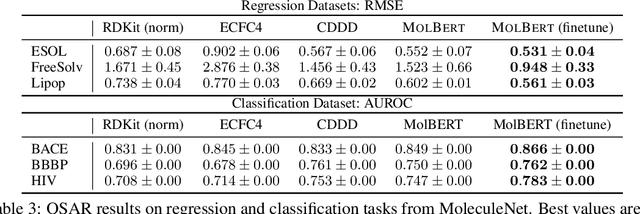
We apply a Transformer architecture, specifically BERT, to learn flexible and high quality molecular representations for drug discovery problems. We study the impact of using different combinations of self-supervised tasks for pre-training, and present our results for the established Virtual Screening and QSAR benchmarks. We show that: i) The selection of appropriate self-supervised task(s) for pre-training has a significant impact on performance in subsequent downstream tasks such as Virtual Screening. ii) Using auxiliary tasks with more domain relevance for Chemistry, such as learning to predict calculated molecular properties, increases the fidelity of our learnt representations. iii) Finally, we show that molecular representations learnt by our model `MolBert' improve upon the current state of the art on the benchmark datasets.
GuacaMol: Benchmarking Models for De Novo Molecular Design
Nov 22, 2018
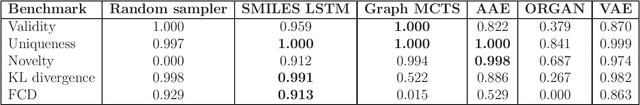
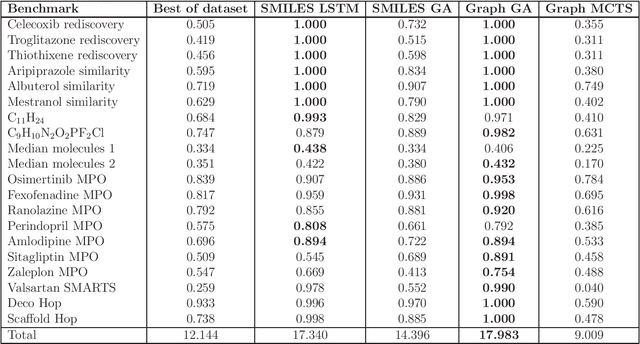
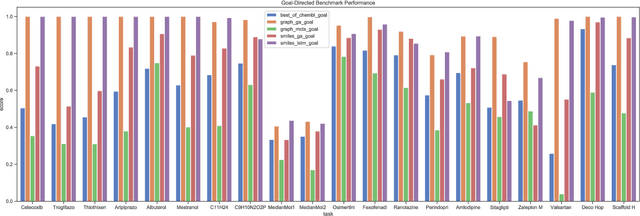
De novo design seeks to generate molecules with required property profiles by virtual design-make-test cycles. With the emergence of deep learning and neural generative models in many application areas, models for molecular design based on neural networks appeared recently and show promising results. However, the new models have not been profiled on consistent tasks, and comparative studies to well-established algorithms have only seldom been performed. To standardize the assessment of both classical and neural models for de novo molecular design, we propose an evaluation framework, GuacaMol, based on a suite of standardized benchmarks. The benchmark tasks encompass measuring the fidelity of the models to reproduce the property distribution of the training sets, the ability to generate novel molecules, the exploration and exploitation of chemical space, and a variety of single and multi-objective optimization tasks. The benchmarking framework is available as an open-source Python package.