 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Scientific Reasoning Model for Organic Synthesis Procedure Generation
Dec 15, 2025Solving computer-aided synthesis planning is essential for enabling fully automated, robot-assisted synthesis workflows and improving the efficiency of drug discovery. A key challenge, however, is bridging the gap between computational route design and practical laboratory execution, particularly the accurate prediction of viable experimental procedures for each synthesis step. In this work, we present QFANG, a scientific reasoning language model capable of generating precise, structured experimental procedures directly from reaction equations, with explicit chain-of-thought reasoning. To develop QFANG, we curated a high-quality dataset comprising 905,990 chemical reactions paired with structured action sequences, extracted and processed from patent literature using large language models. We introduce a Chemistry-Guided Reasoning (CGR) framework that produces chain-of-thought data grounded in chemical knowledge at scale. The model subsequently undergoes supervised fine-tuning to elicit complex chemistry reasoning. Finally, we apply Reinforcement Learning from Verifiable Rewards (RLVR) to further enhance procedural accuracy. Experimental results demonstrate that QFANG outperforms advanced general-purpose reasoning models and nearest-neighbor retrieval baselines, measured by traditional NLP similarity metrics and a chemically aware evaluator using an LLM-as-a-judge. Moreover, QFANG generalizes to certain out-of-domain reaction classes and adapts to variations in laboratory conditions and user-specific constraints. We believe that QFANG's ability to generate high-quality synthesis procedures represents an important step toward bridging the gap between computational synthesis planning and fully automated laboratory synthesis.
UniGenX: Unified Generation of Sequence and Structure with Autoregressive Diffusion
Mar 09, 2025Unified generation of sequence and structure for scientific data (e.g., materials, molecules, proteins) is a critical task. Existing approaches primarily rely on either autoregressive sequence models or diffusion models, each offering distinct advantages and facing notable limitations. Autoregressive models, such as GPT, Llama, and Phi-4, have demonstrated remarkable success in natural language generation and have been extended to multimodal tasks (e.g., image, video, and audio) using advanced encoders like VQ-VAE to represent complex modalities as discrete sequences. However, their direct application to scientific domains is challenging due to the high precision requirements and the diverse nature of scientific data. On the other hand, diffusion models excel at generating high-dimensional scientific data, such as protein, molecule, and material structures, with remarkable accuracy. Yet, their inability to effectively model sequences limits their potential as general-purpose multimodal foundation models. To address these challenges, we propose UniGenX, a unified framework that combines autoregressive next-token prediction with conditional diffusion models. This integration leverages the strengths of autoregressive models to ease the training of conditional diffusion models, while diffusion-based generative heads enhance the precision of autoregressive predictions. We validate the effectiveness of UniGenX on material and small molecule generation tasks, achieving a significant leap in state-of-the-art performance for material crystal structure prediction and establishing new state-of-the-art results for small molecule structure prediction, de novo design, and conditional generation. Notably, UniGenX demonstrates significant improvements, especially in handling long sequences for complex structures, showcasing its efficacy as a versatile tool for scientific data generation.
NatureLM: Deciphering the Language of Nature for Scientific Discovery
Feb 11, 2025



Foundation models have revolutionized natural language processing and artificial intelligence, significantly enhancing how machines comprehend and generate human languages. Inspired by the success of these foundation models, researchers have developed foundation models for individual scientific domains, including small molecules, materials, proteins, DNA, and RNA. However, these models are typically trained in isolation, lacking the ability to integrate across different scientific domains. Recognizing that entities within these domains can all be represented as sequences, which together form the "language of nature", we introduce Nature Language Model (briefly, NatureLM), a sequence-based science foundation model designed for scientific discovery. Pre-trained with data from multiple scientific domains, NatureLM offers a unified, versatile model that enables various applications including: (i) generating and optimizing small molecules, proteins, RNA, and materials using text instructions; (ii) cross-domain generation/design, such as protein-to-molecule and protein-to-RNA generation; and (iii) achieving state-of-the-art performance in tasks like SMILES-to-IUPAC translation and retrosynthesis on USPTO-50k. NatureLM offers a promising generalist approach for various scientific tasks, including drug discovery (hit generation/optimization, ADMET optimization, synthesis), novel material design, and the development of therapeutic proteins or nucleotides. We have developed NatureLM models in different sizes (1 billion, 8 billion, and 46.7 billion parameters) and observed a clear improvement in performance as the model size increases.
Chimera: Accurate retrosynthesis prediction by ensembling models with diverse inductive biases
Dec 06, 2024



Planning and conducting chemical syntheses remains a major bottleneck in the discovery of functional small molecules, and prevents fully leveraging generative AI for molecular inverse design. While early work has shown that ML-based retrosynthesis models can predict reasonable routes, their low accuracy for less frequent, yet important reactions has been pointed out. As multi-step search algorithms are limited to reactions suggested by the underlying model, the applicability of those tools is inherently constrained by the accuracy of retrosynthesis prediction. Inspired by how chemists use different strategies to ideate reactions, we propose Chimera: a framework for building highly accurate reaction models that combine predictions from diverse sources with complementary inductive biases using a learning-based ensembling strategy. We instantiate the framework with two newly developed models, which already by themselves achieve state of the art in their categories. Through experiments across several orders of magnitude in data scale and time-splits, we show Chimera outperforms all major models by a large margin, owing both to the good individual performance of its constituents, but also to the scalability of our ensembling strategy. Moreover, we find that PhD-level organic chemists prefer predictions from Chimera over baselines in terms of quality. Finally, we transfer the largest-scale checkpoint to an internal dataset from a major pharmaceutical company, showing robust generalization under distribution shift. With the new dimension that our framework unlocks, we anticipate further acceleration in the development of even more accurate models.
RetroGFN: Diverse and Feasible Retrosynthesis using GFlowNets
Jun 26, 2024Single-step retrosynthesis aims to predict a set of reactions that lead to the creation of a target molecule, which is a crucial task in molecular discovery. Although a target molecule can often be synthesized with multiple different reactions, it is not clear how to verify the feasibility of a reaction, because the available datasets cover only a tiny fraction of the possible solutions. Consequently, the existing models are not encouraged to explore the space of possible reactions sufficiently. In this paper, we propose a novel single-step retrosynthesis model, RetroGFN, that can explore outside the limited dataset and return a diverse set of feasible reactions by leveraging a feasibility proxy model during the training. We show that RetroGFN achieves competitive results on standard top-k accuracy while outperforming existing methods on round-trip accuracy. Moreover, we provide empirical arguments in favor of using round-trip accuracy which expands the notion of feasibility with respect to the standard top-k accuracy metric.
Generative Active Learning for the Search of Small-molecule Protein Binders
May 02, 2024



Despite substantial progress in machine learning for scientific discovery in recent years, truly de novo design of small molecules which exhibit a property of interest remains a significant challenge. We introduce LambdaZero, a generative active learning approach to search for synthesizable molecules. Powered by deep reinforcement learning, LambdaZero learns to search over the vast space of molecules to discover candidates with a desired property. We apply LambdaZero with molecular docking to design novel small molecules that inhibit the enzyme soluble Epoxide Hydrolase 2 (sEH), while enforcing constraints on synthesizability and drug-likeliness. LambdaZero provides an exponential speedup in terms of the number of calls to the expensive molecular docking oracle, and LambdaZero de novo designed molecules reach docking scores that would otherwise require the virtual screening of a hundred billion molecules. Importantly, LambdaZero discovers novel scaffolds of synthesizable, drug-like inhibitors for sEH. In in vitro experimental validation, a series of ligands from a generated quinazoline-based scaffold were synthesized, and the lead inhibitor N-(4,6-di(pyrrolidin-1-yl)quinazolin-2-yl)-N-methylbenzamide (UM0152893) displayed sub-micromolar enzyme inhibition of sEH.
Re-evaluating Retrosynthesis Algorithms with Syntheseus
Oct 30, 2023The planning of how to synthesize molecules, also known as retrosynthesis, has been a growing focus of the machine learning and chemistry communities in recent years. Despite the appearance of steady progress, we argue that imperfect benchmarks and inconsistent comparisons mask systematic shortcomings of existing techniques. To remedy this, we present a benchmarking library called syntheseus which promotes best practice by default, enabling consistent meaningful evaluation of single-step and multi-step retrosynthesis algorithms. We use syntheseus to re-evaluate a number of previous retrosynthesis algorithms, and find that the ranking of state-of-the-art models changes when evaluated carefully. We end with guidance for future works in this area.
Retro-fallback: retrosynthetic planning in an uncertain world
Oct 13, 2023
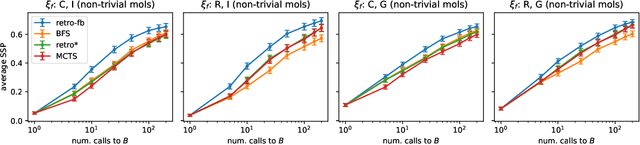


Retrosynthesis is the task of proposing a series of chemical reactions to create a desired molecule from simpler, buyable molecules. While previous works have proposed algorithms to find optimal solutions for a range of metrics (e.g. shortest, lowest-cost), these works generally overlook the fact that we have imperfect knowledge of the space of possible reactions, meaning plans created by the algorithm may not work in a laboratory. In this paper we propose a novel formulation of retrosynthesis in terms of stochastic processes to account for this uncertainty. We then propose a novel greedy algorithm called retro-fallback which maximizes the probability that at least one synthesis plan can be executed in the lab. Using in-silico benchmarks we demonstrate that retro-fallback generally produces better sets of synthesis plans than the popular MCTS and retro* algorithms.
RetroBridge: Modeling Retrosynthesis with Markov Bridges
Aug 30, 2023



Retrosynthesis planning is a fundamental challenge in chemistry which aims at designing reaction pathways from commercially available starting materials to a target molecule. Each step in multi-step retrosynthesis planning requires accurate prediction of possible precursor molecules given the target molecule and confidence estimates to guide heuristic search algorithms. We model single-step retrosynthesis planning as a distribution learning problem in a discrete state space. First, we introduce the Markov Bridge Model, a generative framework aimed to approximate the dependency between two intractable discrete distributions accessible via a finite sample of coupled data points. Our framework is based on the concept of a Markov bridge, a Markov process pinned at its endpoints. Unlike diffusion-based methods, our Markov Bridge Model does not need a tractable noise distribution as a sampling proxy and directly operates on the input product molecules as samples from the intractable prior distribution. We then address the retrosynthesis planning problem with our novel framework and introduce RetroBridge, a template-free retrosynthesis modeling approach that achieves state-of-the-art results on standard evaluation benchmarks.
Are VAEs Bad at Reconstructing Molecular Graphs?
May 04, 2023Many contemporary generative models of molecules are variational auto-encoders of molecular graphs. One term in their training loss pertains to reconstructing the input, yet reconstruction capabilities of state-of-the-art models have not yet been thoroughly compared on a large and chemically diverse dataset. In this work, we show that when several state-of-the-art generative models are evaluated under the same conditions, their reconstruction accuracy is surprisingly low, worse than what was previously reported on seemingly harder datasets. However, we show that improving reconstruction does not directly lead to better sampling or optimization performance. Failed reconstructions from the MoLeR model are usually similar to the inputs, assembling the same motifs in a different way, and possess similar chemical properties such as solubility. Finally, we show that the input molecule and its failed reconstruction are usually mapped by the different encoders to statistically distinguishable posterior distributions, hinting that posterior collapse may not fully explain why VAEs are bad at reconstructing molecular graphs.