 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAre VAEs Bad at Reconstructing Molecular Graphs?
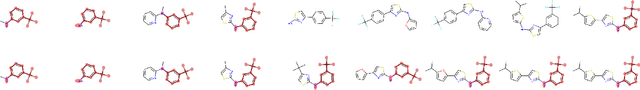
May 04, 2023Many contemporary generative models of molecules are variational auto-encoders of molecular graphs. One term in their training loss pertains to reconstructing the input, yet reconstruction capabilities of state-of-the-art models have not yet been thoroughly compared on a large and chemically diverse dataset. In this work, we show that when several state-of-the-art generative models are evaluated under the same conditions, their reconstruction accuracy is surprisingly low, worse than what was previously reported on seemingly harder datasets. However, we show that improving reconstruction does not directly lead to better sampling or optimization performance. Failed reconstructions from the MoLeR model are usually similar to the inputs, assembling the same motifs in a different way, and possess similar chemical properties such as solubility. Finally, we show that the input molecule and its failed reconstruction are usually mapped by the different encoders to statistically distinguishable posterior distributions, hinting that posterior collapse may not fully explain why VAEs are bad at reconstructing molecular graphs.
Learning to Extend Molecular Scaffolds with Structural Motifs
Mar 05, 2021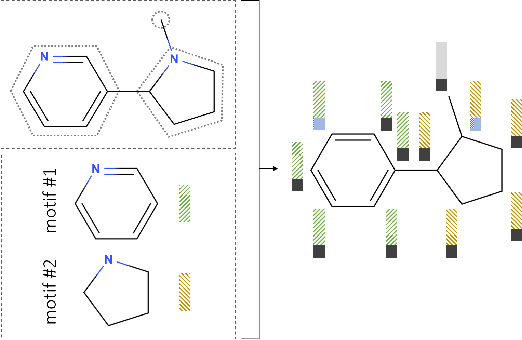

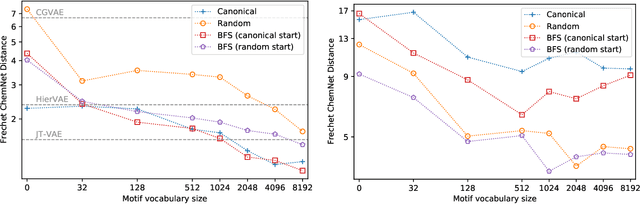
Recent advancements in deep learning-based modeling of molecules promise to accelerate in silico drug discovery. There is a plethora of generative models available, which build molecules either atom-by-atom and bond-by-bond or fragment-by-fragment. Many drug discovery projects also require a fixed scaffold to be present in the generated molecule, and incorporating that constraint has been recently explored. In this work, we propose a new graph-based model that learns to extend a given partial molecule by flexibly choosing between adding individual atoms and entire fragments. Extending a scaffold is implemented by using it as the initial partial graph, which is possible because our model does not depend on generation history. We show that training using a randomized generation order is necessary for good performance when extending scaffolds, and that the results are further improved by increasing fragment vocabulary size. Our model pushes the state-of-the-art of graph-based molecule generation, while being an order of magnitude faster to train and sample from than existing approaches.