 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMolecularIQ: Characterizing Chemical Reasoning Capabilities Through Symbolic Verification on Molecular Graphs
Jan 21, 2026A molecule's properties are fundamentally determined by its composition and structure encoded in its molecular graph. Thus, reasoning about molecular properties requires the ability to parse and understand the molecular graph. Large Language Models (LLMs) are increasingly applied to chemistry, tackling tasks such as molecular name conversion, captioning, text-guided generation, and property or reaction prediction. Most existing benchmarks emphasize general chemical knowledge, rely on literature or surrogate labels that risk leakage or bias, or reduce evaluation to multiple-choice questions. We introduce MolecularIQ, a molecular structure reasoning benchmark focused exclusively on symbolically verifiable tasks. MolecularIQ enables fine-grained evaluation of reasoning over molecular graphs and reveals capability patterns that localize model failures to specific tasks and molecular structures. This provides actionable insights into the strengths and limitations of current chemistry LLMs and guides the development of models that reason faithfully over molecular structure.
Federated Few-Shot Learning for Epileptic Seizure Detection Under Privacy Constraints
Dec 09, 2025Many deep learning approaches have been developed for EEG-based seizure detection; however, most rely on access to large centralized annotated datasets. In clinical practice, EEG data are often scarce, patient-specific distributed across institutions, and governed by strict privacy regulations that prohibit data pooling. As a result, creating usable AI-based seizure detection models remains challenging in real-world medical settings. To address these constraints, we propose a two-stage federated few-shot learning (FFSL) framework for personalized EEG-based seizure detection. The method is trained and evaluated on the TUH Event Corpus, which includes six EEG event classes. In Stage 1, a pretrained biosignal transformer (BIOT) is fine-tuned across non-IID simulated hospital sites using federated learning, enabling shared representation learning without centralizing EEG recordings. In Stage 2, federated few-shot personalization adapts the classifier to each patient using only five labeled EEG segments, retaining seizure-specific information while still benefiting from cross-site knowledge. Federated fine-tuning achieved a balanced accuracy of 0.43 (centralized: 0.52), Cohen's kappa of 0.42 (0.49), and weighted F1 of 0.69 (0.74). In the FFSL stage, client-specific models reached an average balanced accuracy of 0.77, Cohen's kappa of 0.62, and weighted F1 of 0.73 across four sites with heterogeneous event distributions. These results suggest that FFSL can support effective patient-adaptive seizure detection under realistic data-availability and privacy constraints.
Bio-xLSTM: Generative modeling, representation and in-context learning of biological and chemical sequences
Nov 06, 2024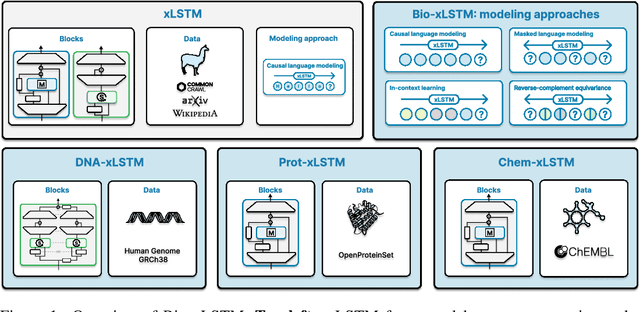
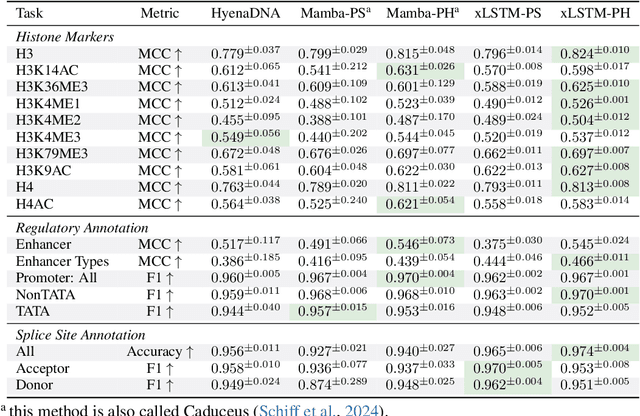
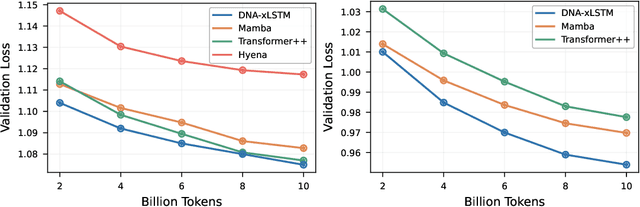
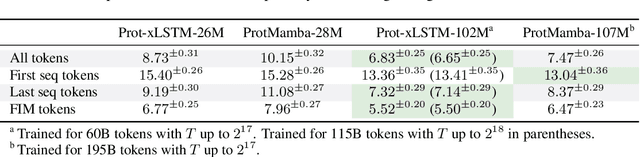
Language models for biological and chemical sequences enable crucial applications such as drug discovery, protein engineering, and precision medicine. Currently, these language models are predominantly based on Transformer architectures. While Transformers have yielded impressive results, their quadratic runtime dependency on the sequence length complicates their use for long genomic sequences and in-context learning on proteins and chemical sequences. Recently, the recurrent xLSTM architecture has been shown to perform favorably compared to Transformers and modern state-space model (SSM) architectures in the natural language domain. Similar to SSMs, xLSTMs have a linear runtime dependency on the sequence length and allow for constant-memory decoding at inference time, which makes them prime candidates for modeling long-range dependencies in biological and chemical sequences. In this work, we tailor xLSTM towards these domains and propose a suite of architectural variants called Bio-xLSTM. Extensive experiments in three large domains, genomics, proteins, and chemistry, were performed to assess xLSTM's ability to model biological and chemical sequences. The results show that models based on Bio-xLSTM a) can serve as proficient generative models for DNA, protein, and chemical sequences, b) learn rich representations for those modalities, and c) can perform in-context learning for proteins and small molecules.
Re-evaluating Retrosynthesis Algorithms with Syntheseus
Oct 30, 2023The planning of how to synthesize molecules, also known as retrosynthesis, has been a growing focus of the machine learning and chemistry communities in recent years. Despite the appearance of steady progress, we argue that imperfect benchmarks and inconsistent comparisons mask systematic shortcomings of existing techniques. To remedy this, we present a benchmarking library called syntheseus which promotes best practice by default, enabling consistent meaningful evaluation of single-step and multi-step retrosynthesis algorithms. We use syntheseus to re-evaluate a number of previous retrosynthesis algorithms, and find that the ranking of state-of-the-art models changes when evaluated carefully. We end with guidance for future works in this area.
Enhancing Activity Prediction Models in Drug Discovery with the Ability to Understand Human Language
Mar 06, 2023Activity and property prediction models are the central workhorses in drug discovery and materials sciences, but currently they have to be trained or fine-tuned for new tasks. Without training or fine-tuning, scientific language models could be used for such low-data tasks through their announced zero- and few-shot capabilities. However, their predictive quality at activity prediction is lacking. In this work, we envision a novel type of activity prediction model that is able to adapt to new prediction tasks at inference time, via understanding textual information describing the task. To this end, we propose a new architecture with separate modules for chemical and natural language inputs, and a contrastive pre-training objective on data from large biochemical databases. In extensive experiments, we show that our method CLAMP yields improved predictive performance on few-shot learning benchmarks and zero-shot problems in drug discovery. We attribute the advances of our method to the modularized architecture and to our pre-training objective.
Supervised machine learning classification for short straddles on the S&P500
Apr 26, 2022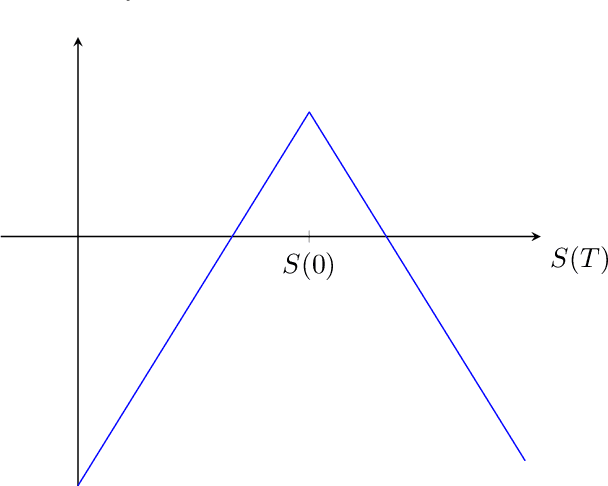

In this working paper we present our current progress in the training of machine learning models to execute short option strategies on the S&P500. As a first step, this paper is breaking this problem down to a supervised classification task to decide if a short straddle on the S&P500 should be executed or not on a daily basis. We describe our used framework and present an overview over our evaluation metrics on different classification models. In this preliminary work, using standard machine learning techniques and without hyperparameter search, we find no statistically significant outperformance to a simple "trade always" strategy, but gain additional insights on how we could proceed in further experiments.
Modern Hopfield Networks for Few- and Zero-Shot Reaction Prediction
Apr 07, 2021
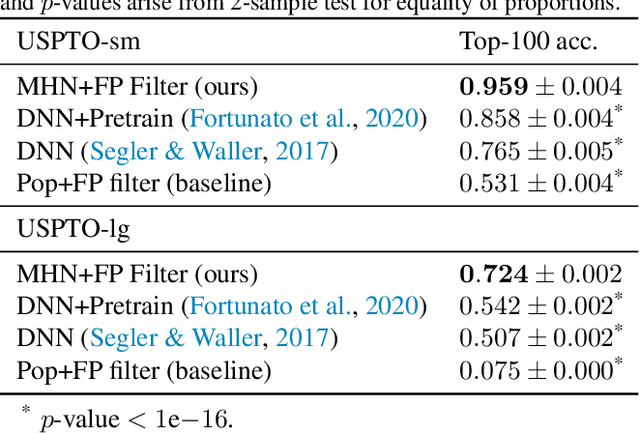
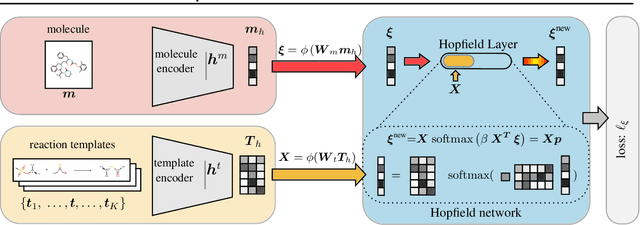
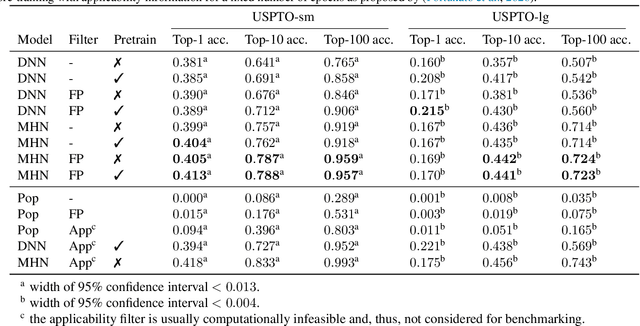
An essential step in the discovery of new drugs and materials is the synthesis of a molecule that exists so far only as an idea to test its biological and physical properties. While computer-aided design of virtual molecules has made large progress, computer-assisted synthesis planning (CASP) to realize physical molecules is still in its infancy and lacks a performance level that would enable large-scale molecule discovery. CASP supports the search for multi-step synthesis routes, which is very challenging due to high branching factors in each synthesis step and the hidden rules that govern the reactions. The central and repeatedly applied step in CASP is reaction prediction, for which machine learning methods yield the best performance. We propose a novel reaction prediction approach that uses a deep learning architecture with modern Hopfield networks (MHNs) that is optimized by contrastive learning. An MHN is an associative memory that can store and retrieve chemical reactions in each layer of a deep learning architecture. We show that our MHN contrastive learning approach enables few- and zero-shot learning for reaction prediction which, in contrast to previous methods, can deal with rare, single, or even no training example(s) for a reaction. On a well established benchmark, our MHN approach pushes the state-of-the-art performance up by a large margin as it improves the predictive top-100 accuracy from $0.858\pm0.004$ to $0.959\pm0.004$. This advance might pave the way to large-scale molecule discovery.
Hopfield Networks is All You Need
Jul 16, 2020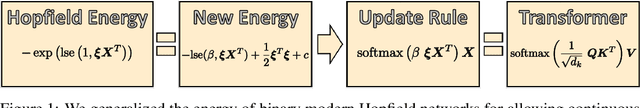
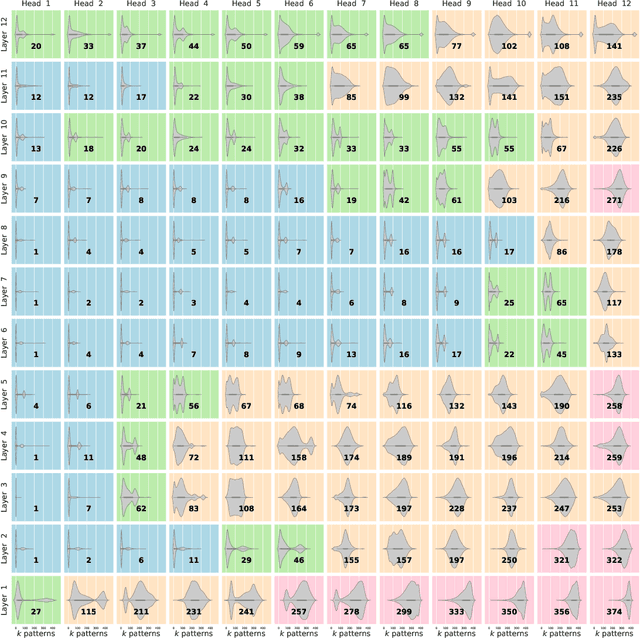
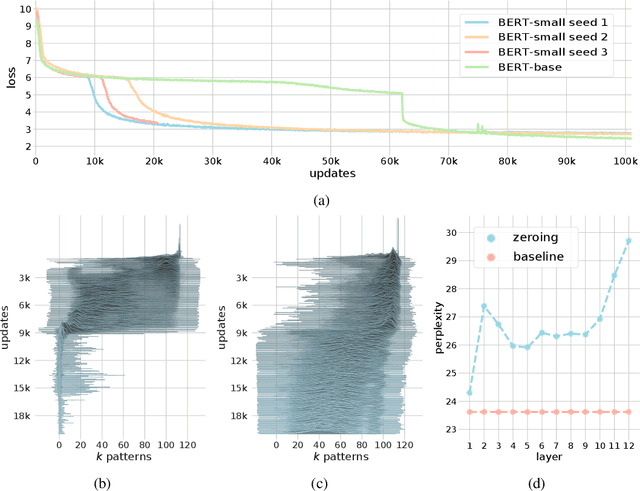
We show that the transformer attention mechanism is the update rule of a modern Hopfield network with continuous states. This new Hopfield network can store exponentially (with the dimension) many patterns, converges with one update, and has exponentially small retrieval errors. The number of stored patterns is traded off against convergence speed and retrieval error. The new Hopfield network has three types of energy minima (fixed points of the update): (1) global fixed point averaging over all patterns, (2) metastable states averaging over a subset of patterns, and (3) fixed points which store a single pattern. Transformer and BERT models operate in their first layers preferably in the global averaging regime, while they operate in higher layers in metastable states. The gradient in transformers is maximal for metastable states, is uniformly distributed for global averaging, and vanishes for a fixed point near a stored pattern. Using the Hopfield network interpretation, we analyzed learning of transformer and BERT models. Learning starts with attention heads that average and then most of them switch to metastable states. However, the majority of heads in the first layers still averages and can be replaced by averaging, e.g. our proposed Gaussian weighting. In contrast, heads in the last layers steadily learn and seem to use metastable states to collect information created in lower layers. These heads seem to be a promising target for improving transformers. Neural networks with Hopfield networks outperform other methods on immune repertoire classification, where the Hopfield net stores several hundreds of thousands of patterns. We provide a new PyTorch layer called "Hopfield", which allows to equip deep learning architectures with modern Hopfield networks as a new powerful concept comprising pooling, memory, and attention. GitHub: https://github.com/ml-jku/hopfield-layers
Large-scale ligand-based virtual screening for SARS-CoV-2 inhibitors using deep neural networks
Apr 03, 2020
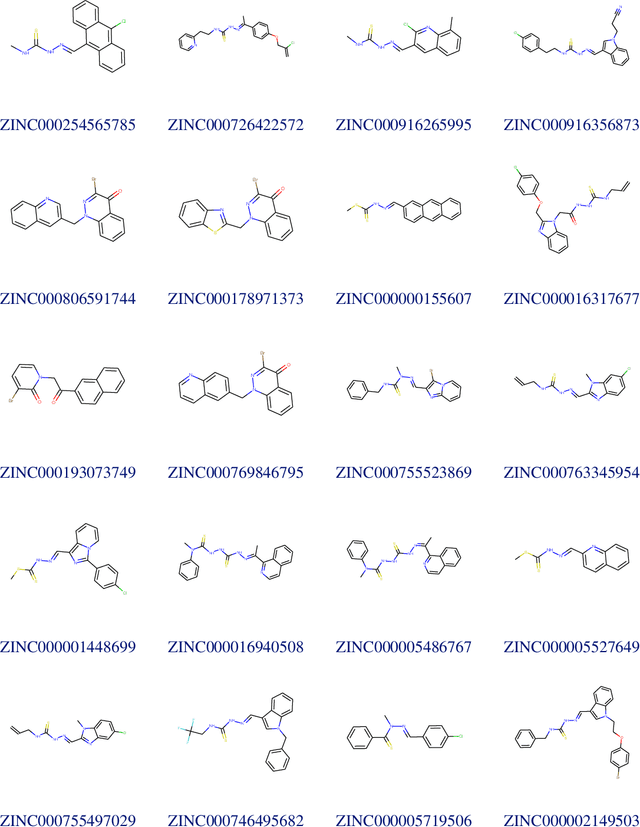

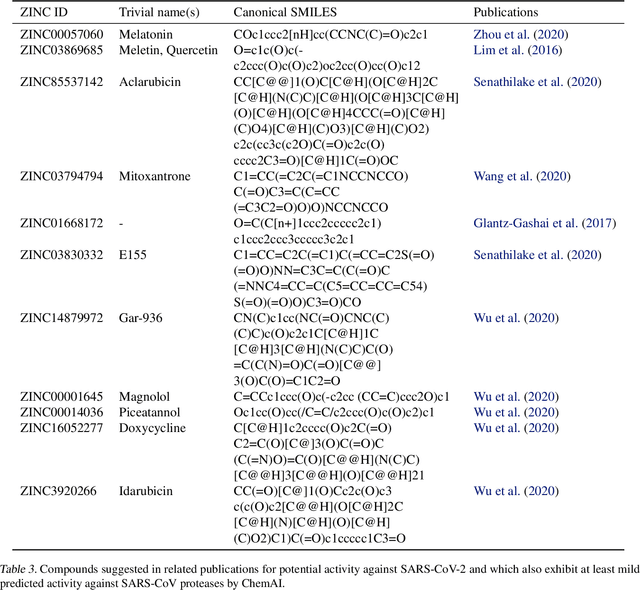
Due to the current severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic, there is an urgent need for novel therapies and drugs. We conducted a large-scale virtual screening for small molecules that are potential CoV-2 inhibitors. To this end, we utilized "ChemAI", a deep neural network trained on more than 220M data points across 3.6M molecules from three public drug-discovery databases. With ChemAI, we screened and ranked one billion molecules from the ZINC database for favourable effects against CoV-2. We then reduced the result to the 30,000 top-ranked compounds, which are readily accessible and purchasable via the ZINC database. Additionally, we screened the DrugBank using ChemAI to allow for drug repurposing, which would be a fast way towards a therapy. We provide these top-ranked compounds of ZINC and DrugBank as a library for further screening with bioassays at https://github.com/ml-jku/sars-cov-inhibitors-chemai.