 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeModels Matter: The Impact of Single-Step Retrosynthesis on Synthesis Planning
Aug 10, 2023



Retrosynthesis consists of breaking down a chemical compound recursively step-by-step into molecular precursors until a set of commercially available molecules is found with the goal to provide a synthesis route. Its two primary research directions, single-step retrosynthesis prediction, which models the chemical reaction logic, and multi-step synthesis planning, which tries to find the correct sequence of reactions, are inherently intertwined. Still, this connection is not reflected in contemporary research. In this work, we combine these two major research directions by applying multiple single-step retrosynthesis models within multi-step synthesis planning and analyzing their impact using public and proprietary reaction data. We find a disconnection between high single-step performance and potential route-finding success, suggesting that single-step models must be evaluated within synthesis planning in the future. Furthermore, we show that the commonly used single-step retrosynthesis benchmark dataset USPTO-50k is insufficient as this evaluation task does not represent model performance and scalability on larger and more diverse datasets. For multi-step synthesis planning, we show that the choice of the single-step model can improve the overall success rate of synthesis planning by up to +28% compared to the commonly used baseline model. Finally, we show that each single-step model finds unique synthesis routes, and differs in aspects such as route-finding success, the number of found synthesis routes, and chemical validity, making the combination of single-step retrosynthesis prediction and multi-step synthesis planning a crucial aspect when developing future methods.
Mind the Retrosynthesis Gap: Bridging the divide between Single-step and Multi-step Retrosynthesis Prediction
Dec 12, 2022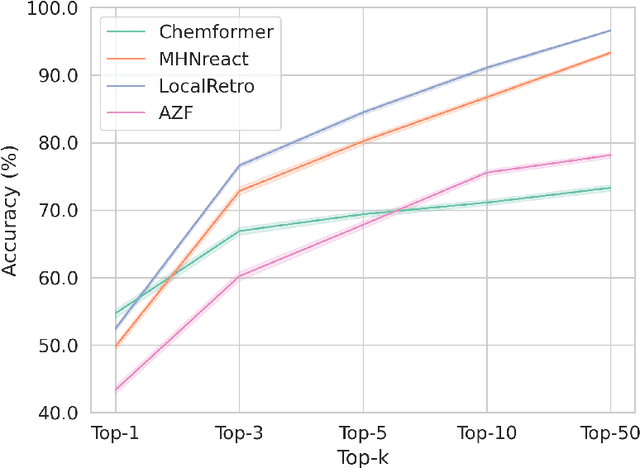
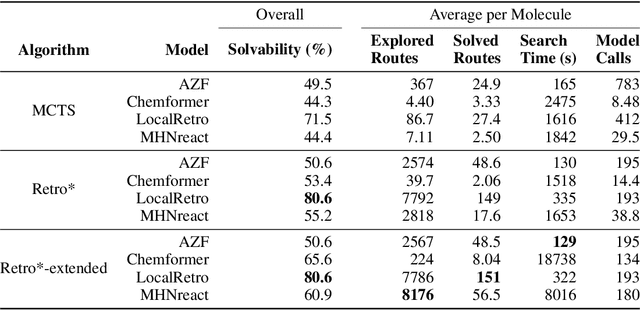
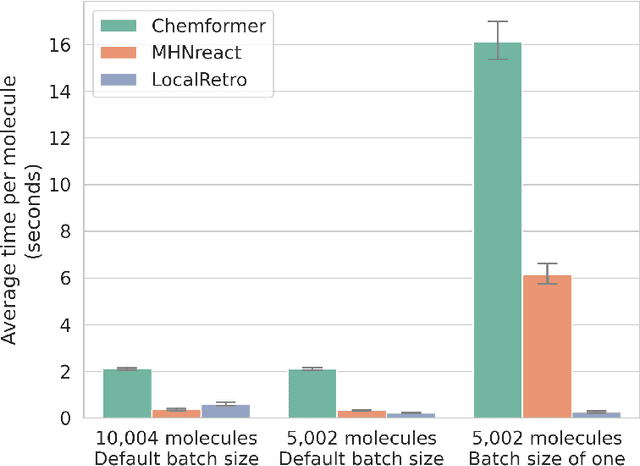
Retrosynthesis is the task of breaking down a chemical compound recursively step-by-step into molecular precursors until a set of commercially available molecules is found. Consequently, the goal is to provide a valid synthesis route for a molecule. As more single-step models develop, we see increasing accuracy in the prediction of molecular disconnections, potentially improving the creation of synthetic paths. Multi-step approaches repeatedly apply the chemical information stored in single-step retrosynthesis models. However, this connection is not reflected in contemporary research, fixing either the single-step model or the multi-step algorithm in the process. In this work, we establish a bridge between both tasks by benchmarking the performance and transfer of different single-step retrosynthesis models to the multi-step domain by leveraging two common search algorithms, Monte Carlo Tree Search and Retro*. We show that models designed for single-step retrosynthesis, when extended to multi-step, can have a tremendous impact on the route finding capabilities of current multi-step methods, improving performance by up to +30% compared to the most widely used model. Furthermore, we observe no clear link between contemporary single-step and multi-step evaluation metrics, showing that single-step models need to be developed and tested for the multi-step domain and not as an isolated task to find synthesis routes for molecules of interest.
Modern Hopfield Networks for Few- and Zero-Shot Reaction Prediction
Apr 07, 2021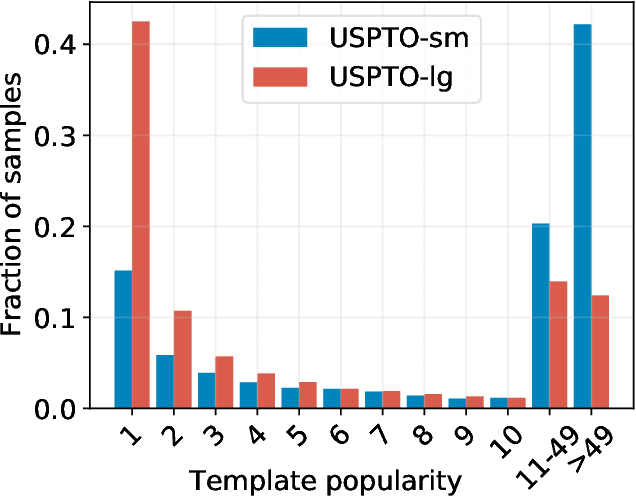
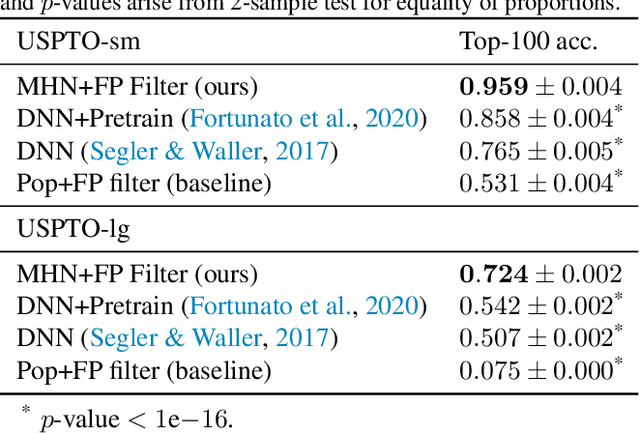
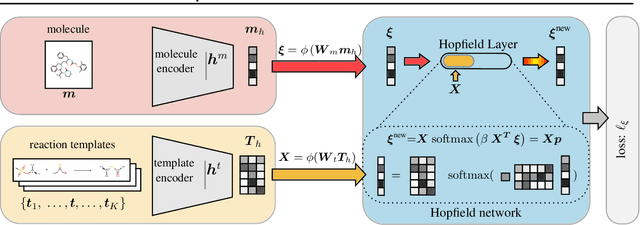
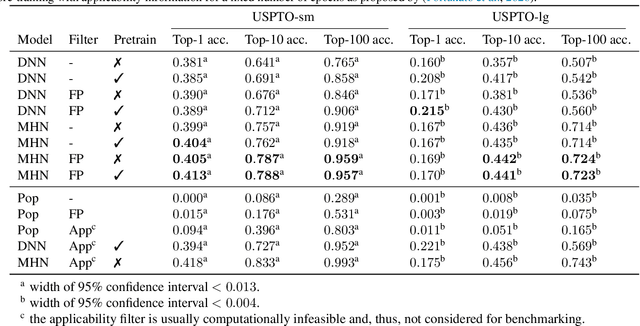
An essential step in the discovery of new drugs and materials is the synthesis of a molecule that exists so far only as an idea to test its biological and physical properties. While computer-aided design of virtual molecules has made large progress, computer-assisted synthesis planning (CASP) to realize physical molecules is still in its infancy and lacks a performance level that would enable large-scale molecule discovery. CASP supports the search for multi-step synthesis routes, which is very challenging due to high branching factors in each synthesis step and the hidden rules that govern the reactions. The central and repeatedly applied step in CASP is reaction prediction, for which machine learning methods yield the best performance. We propose a novel reaction prediction approach that uses a deep learning architecture with modern Hopfield networks (MHNs) that is optimized by contrastive learning. An MHN is an associative memory that can store and retrieve chemical reactions in each layer of a deep learning architecture. We show that our MHN contrastive learning approach enables few- and zero-shot learning for reaction prediction which, in contrast to previous methods, can deal with rare, single, or even no training example(s) for a reaction. On a well established benchmark, our MHN approach pushes the state-of-the-art performance up by a large margin as it improves the predictive top-100 accuracy from $0.858\pm0.004$ to $0.959\pm0.004$. This advance might pave the way to large-scale molecule discovery.