 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEffective Distillation to Hybrid xLSTM Architectures
Mar 16, 2026There have been numerous attempts to distill quadratic attention-based large language models (LLMs) into sub-quadratic linearized architectures. However, despite extensive research, such distilled models often fail to match the performance of their teacher LLMs on various downstream tasks. We set out the goal of lossless distillation, which we define in terms of tolerance-corrected Win-and-Tie rates between student and teacher on sets of tasks. To this end, we introduce an effective distillation pipeline for xLSTM-based students. We propose an additional merging stage, where individually linearized experts are combined into a single model. We show the effectiveness of this pipeline by distilling base and instruction-tuned models from the Llama, Qwen, and Olmo families. In many settings, our xLSTM-based students recover most of the teacher's performance, and even exceed it on some downstream tasks. Our contributions are an important step towards more energy-efficient and cost-effective replacements for transformer-based LLMs.
Symbol-Equivariant Recurrent Reasoning Models
Mar 02, 2026Reasoning problems such as Sudoku and ARC-AGI remain challenging for neural networks. The structured problem solving architecture family of Recurrent Reasoning Models (RRMs), including Hierarchical Reasoning Model (HRM) and Tiny Recursive Model (TRM), offer a compact alternative to large language models, but currently handle symbol symmetries only implicitly via costly data augmentation. We introduce Symbol-Equivariant Recurrent Reasoning Models (SE-RRMs), which enforce permutation equivariance at the architectural level through symbol-equivariant layers, guaranteeing identical solutions under symbol or color permutations. SE-RRMs outperform prior RRMs on 9x9 Sudoku and generalize from just training on 9x9 to smaller 4x4 and larger 16x16 and 25x25 instances, to which existing RRMs cannot extrapolate. On ARC-AGI-1 and ARC-AGI-2, SE-RRMs achieve competitive performance with substantially less data augmentation and only 2 million parameters, demonstrating that explicitly encoding symmetry improves the robustness and scalability of neural reasoning. Code is available at https://github.com/ml-jku/SE-RRM.
MolecularIQ: Characterizing Chemical Reasoning Capabilities Through Symbolic Verification on Molecular Graphs
Jan 21, 2026A molecule's properties are fundamentally determined by its composition and structure encoded in its molecular graph. Thus, reasoning about molecular properties requires the ability to parse and understand the molecular graph. Large Language Models (LLMs) are increasingly applied to chemistry, tackling tasks such as molecular name conversion, captioning, text-guided generation, and property or reaction prediction. Most existing benchmarks emphasize general chemical knowledge, rely on literature or surrogate labels that risk leakage or bias, or reduce evaluation to multiple-choice questions. We introduce MolecularIQ, a molecular structure reasoning benchmark focused exclusively on symbolically verifiable tasks. MolecularIQ enables fine-grained evaluation of reasoning over molecular graphs and reveals capability patterns that localize model failures to specific tasks and molecular structures. This provides actionable insights into the strengths and limitations of current chemistry LLMs and guides the development of models that reason faithfully over molecular structure.
Contrastive Geometric Learning Unlocks Unified Structure- and Ligand-Based Drug Design
Jan 14, 2026Structure-based and ligand-based computational drug design have traditionally relied on disjoint data sources and modeling assumptions, limiting their joint use at scale. In this work, we introduce Contrastive Geometric Learning for Unified Computational Drug Design (ConGLUDe), a single contrastive geometric model that unifies structure- and ligand-based training. ConGLUDe couples a geometric protein encoder that produces whole-protein representations and implicit embeddings of predicted binding sites with a fast ligand encoder, removing the need for pre-defined pockets. By aligning ligands with both global protein representations and multiple candidate binding sites through contrastive learning, ConGLUDe supports ligand-conditioned pocket prediction in addition to virtual screening and target fishing, while being trained jointly on protein-ligand complexes and large-scale bioactivity data. Across diverse benchmarks, ConGLUDe achieves state-of-the-art zero-shot virtual screening performance in settings where no binding pocket information is provided as input, substantially outperforms existing methods on a challenging target fishing task, and demonstrates competitive ligand-conditioned pocket selection. These results highlight the advantages of unified structure-ligand training and position ConGLUDe as a step toward general-purpose foundation models for drug discovery.
Measuring AI Progress in Drug Discovery: A Reproducible Leaderboard for the Tox21 Challenge
Nov 18, 2025Deep learning's rise since the early 2010s has transformed fields like computer vision and natural language processing and strongly influenced biomedical research. For drug discovery specifically, a key inflection - akin to vision's "ImageNet moment" - arrived in 2015, when deep neural networks surpassed traditional approaches on the Tox21 Data Challenge. This milestone accelerated the adoption of deep learning across the pharmaceutical industry, and today most major companies have integrated these methods into their research pipelines. After the Tox21 Challenge concluded, its dataset was included in several established benchmarks, such as MoleculeNet and the Open Graph Benchmark. However, during these integrations, the dataset was altered and labels were imputed or manufactured, resulting in a loss of comparability across studies. Consequently, the extent to which bioactivity and toxicity prediction methods have improved over the past decade remains unclear. To this end, we introduce a reproducible leaderboard, hosted on Hugging Face with the original Tox21 Challenge dataset, together with a set of baseline and representative methods. The current version of the leaderboard indicates that the original Tox21 winner - the ensemble-based DeepTox method - and the descriptor-based self-normalizing neural networks introduced in 2017, continue to perform competitively and rank among the top methods for toxicity prediction, leaving it unclear whether substantial progress in toxicity prediction has been achieved over the past decade. As part of this work, we make all baselines and evaluated models publicly accessible for inference via standardized API calls to Hugging Face Spaces.
TiRex: Zero-Shot Forecasting Across Long and Short Horizons with Enhanced In-Context Learning
May 29, 2025In-context learning, the ability of large language models to perform tasks using only examples provided in the prompt, has recently been adapted for time series forecasting. This paradigm enables zero-shot prediction, where past values serve as context for forecasting future values, making powerful forecasting tools accessible to non-experts and increasing the performance when training data are scarce. Most existing zero-shot forecasting approaches rely on transformer architectures, which, despite their success in language, often fall short of expectations in time series forecasting, where recurrent models like LSTMs frequently have the edge. Conversely, while LSTMs are well-suited for time series modeling due to their state-tracking capabilities, they lack strong in-context learning abilities. We introduce TiRex that closes this gap by leveraging xLSTM, an enhanced LSTM with competitive in-context learning skills. Unlike transformers, state-space models, or parallelizable RNNs such as RWKV, TiRex retains state-tracking, a critical property for long-horizon forecasting. To further facilitate its state-tracking ability, we propose a training-time masking strategy called CPM. TiRex sets a new state of the art in zero-shot time series forecasting on the HuggingFace benchmarks GiftEval and Chronos-ZS, outperforming significantly larger models including TabPFN-TS (Prior Labs), Chronos Bolt (Amazon), TimesFM (Google), and Moirai (Salesforce) across both short- and long-term forecasts.
xLSTM 7B: A Recurrent LLM for Fast and Efficient Inference
Mar 17, 2025Recent breakthroughs in solving reasoning, math and coding problems with Large Language Models (LLMs) have been enabled by investing substantial computation budgets at inference time. Therefore, inference speed is one of the most critical properties of LLM architectures, and there is a growing need for LLMs that are efficient and fast at inference. Recently, LLMs built on the xLSTM architecture have emerged as a powerful alternative to Transformers, offering linear compute scaling with sequence length and constant memory usage, both highly desirable properties for efficient inference. However, such xLSTM-based LLMs have yet to be scaled to larger models and assessed and compared with respect to inference speed and efficiency. In this work, we introduce xLSTM 7B, a 7-billion-parameter LLM that combines xLSTM's architectural benefits with targeted optimizations for fast and efficient inference. Our experiments demonstrate that xLSTM 7B achieves performance on downstream tasks comparable to other similar-sized LLMs, while providing significantly faster inference speeds and greater efficiency compared to Llama- and Mamba-based LLMs. These results establish xLSTM 7B as the fastest and most efficient 7B LLM, offering a solution for tasks that require large amounts of test-time computation. Our work highlights xLSTM's potential as a foundational architecture for methods building on heavy use of LLM inference. Our model weights, model code and training code are open-source.
LaM-SLidE: Latent Space Modeling of Spatial Dynamical Systems via Linked Entities
Feb 17, 2025Generative models are spearheading recent progress in deep learning, showing strong promise for trajectory sampling in dynamical systems as well. However, while latent space modeling paradigms have transformed image and video generation, similar approaches are more difficult for most dynamical systems. Such systems -- from chemical molecule structures to collective human behavior -- are described by interactions of entities, making them inherently linked to connectivity patterns and the traceability of entities over time. Our approach, LaM-SLidE (Latent Space Modeling of Spatial Dynamical Systems via Linked Entities), combines the advantages of graph neural networks, i.e., the traceability of entities across time-steps, with the efficiency and scalability of recent advances in image and video generation, where pre-trained encoder and decoder are frozen to enable generative modeling in the latent space. The core idea of LaM-SLidE is to introduce identifier representations (IDs) to allow for retrieval of entity properties, e.g., entity coordinates, from latent system representations and thus enables traceability. Experimentally, across different domains, we show that LaM-SLidE performs favorably in terms of speed, accuracy, and generalizability. (Code is available at https://github.com/ml-jku/LaM-SLidE)
Bio-xLSTM: Generative modeling, representation and in-context learning of biological and chemical sequences
Nov 06, 2024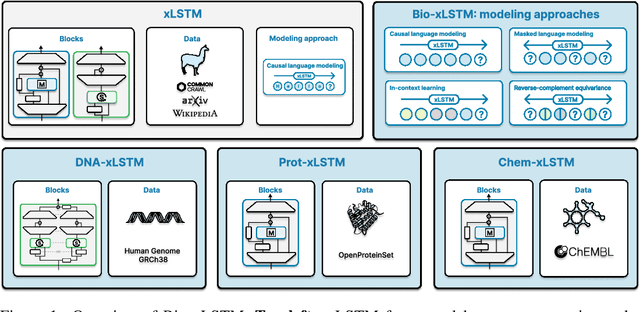
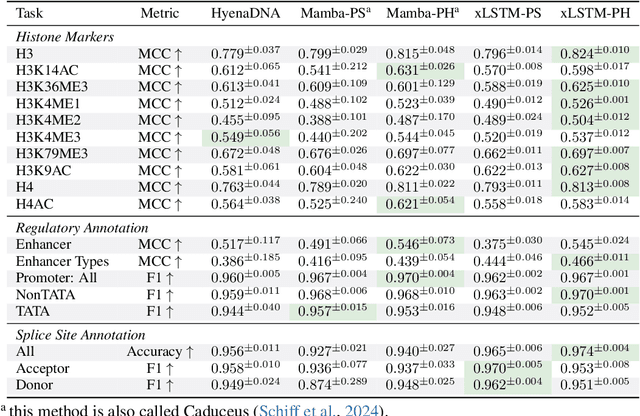
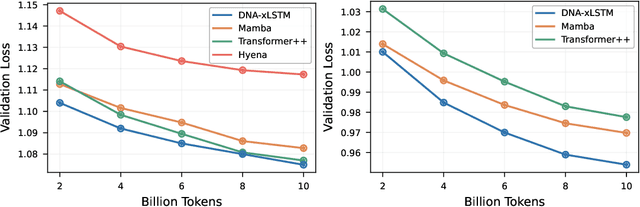
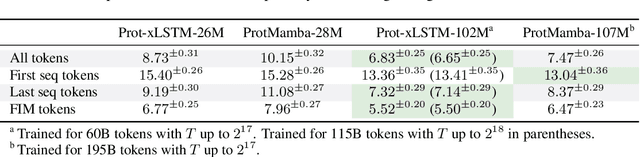
Language models for biological and chemical sequences enable crucial applications such as drug discovery, protein engineering, and precision medicine. Currently, these language models are predominantly based on Transformer architectures. While Transformers have yielded impressive results, their quadratic runtime dependency on the sequence length complicates their use for long genomic sequences and in-context learning on proteins and chemical sequences. Recently, the recurrent xLSTM architecture has been shown to perform favorably compared to Transformers and modern state-space model (SSM) architectures in the natural language domain. Similar to SSMs, xLSTMs have a linear runtime dependency on the sequence length and allow for constant-memory decoding at inference time, which makes them prime candidates for modeling long-range dependencies in biological and chemical sequences. In this work, we tailor xLSTM towards these domains and propose a suite of architectural variants called Bio-xLSTM. Extensive experiments in three large domains, genomics, proteins, and chemistry, were performed to assess xLSTM's ability to model biological and chemical sequences. The results show that models based on Bio-xLSTM a) can serve as proficient generative models for DNA, protein, and chemical sequences, b) learn rich representations for those modalities, and c) can perform in-context learning for proteins and small molecules.
A Large Recurrent Action Model: xLSTM enables Fast Inference for Robotics Tasks
Oct 29, 2024In recent years, there has been a trend in the field of Reinforcement Learning (RL) towards large action models trained offline on large-scale datasets via sequence modeling. Existing models are primarily based on the Transformer architecture, which result in powerful agents. However, due to slow inference times, Transformer-based approaches are impractical for real-time applications, such as robotics. Recently, modern recurrent architectures, such as xLSTM and Mamba, have been proposed that exhibit parallelization benefits during training similar to the Transformer architecture while offering fast inference. In this work, we study the aptitude of these modern recurrent architectures for large action models. Consequently, we propose a Large Recurrent Action Model (LRAM) with an xLSTM at its core that comes with linear-time inference complexity and natural sequence length extrapolation abilities. Experiments on 432 tasks from 6 domains show that LRAM compares favorably to Transformers in terms of performance and speed.