 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSymbol-Equivariant Recurrent Reasoning Models
Mar 02, 2026Reasoning problems such as Sudoku and ARC-AGI remain challenging for neural networks. The structured problem solving architecture family of Recurrent Reasoning Models (RRMs), including Hierarchical Reasoning Model (HRM) and Tiny Recursive Model (TRM), offer a compact alternative to large language models, but currently handle symbol symmetries only implicitly via costly data augmentation. We introduce Symbol-Equivariant Recurrent Reasoning Models (SE-RRMs), which enforce permutation equivariance at the architectural level through symbol-equivariant layers, guaranteeing identical solutions under symbol or color permutations. SE-RRMs outperform prior RRMs on 9x9 Sudoku and generalize from just training on 9x9 to smaller 4x4 and larger 16x16 and 25x25 instances, to which existing RRMs cannot extrapolate. On ARC-AGI-1 and ARC-AGI-2, SE-RRMs achieve competitive performance with substantially less data augmentation and only 2 million parameters, demonstrating that explicitly encoding symmetry improves the robustness and scalability of neural reasoning. Code is available at https://github.com/ml-jku/SE-RRM.
LaM-SLidE: Latent Space Modeling of Spatial Dynamical Systems via Linked Entities
Feb 17, 2025Generative models are spearheading recent progress in deep learning, showing strong promise for trajectory sampling in dynamical systems as well. However, while latent space modeling paradigms have transformed image and video generation, similar approaches are more difficult for most dynamical systems. Such systems -- from chemical molecule structures to collective human behavior -- are described by interactions of entities, making them inherently linked to connectivity patterns and the traceability of entities over time. Our approach, LaM-SLidE (Latent Space Modeling of Spatial Dynamical Systems via Linked Entities), combines the advantages of graph neural networks, i.e., the traceability of entities across time-steps, with the efficiency and scalability of recent advances in image and video generation, where pre-trained encoder and decoder are frozen to enable generative modeling in the latent space. The core idea of LaM-SLidE is to introduce identifier representations (IDs) to allow for retrieval of entity properties, e.g., entity coordinates, from latent system representations and thus enables traceability. Experimentally, across different domains, we show that LaM-SLidE performs favorably in terms of speed, accuracy, and generalizability. (Code is available at https://github.com/ml-jku/LaM-SLidE)
Bio-xLSTM: Generative modeling, representation and in-context learning of biological and chemical sequences
Nov 06, 2024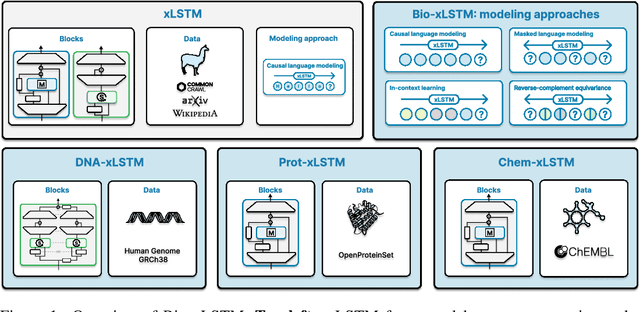
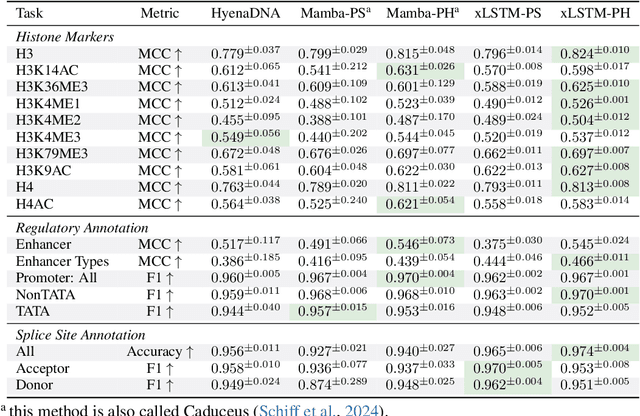
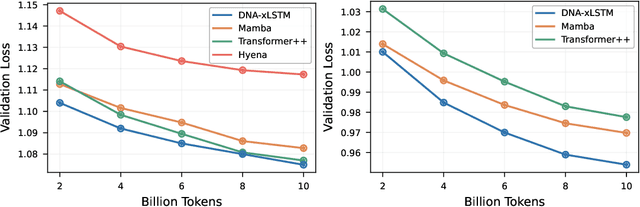
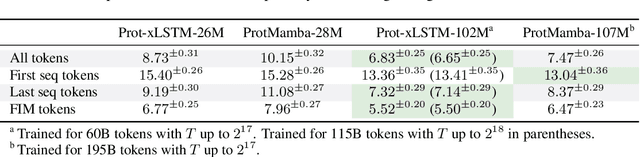
Language models for biological and chemical sequences enable crucial applications such as drug discovery, protein engineering, and precision medicine. Currently, these language models are predominantly based on Transformer architectures. While Transformers have yielded impressive results, their quadratic runtime dependency on the sequence length complicates their use for long genomic sequences and in-context learning on proteins and chemical sequences. Recently, the recurrent xLSTM architecture has been shown to perform favorably compared to Transformers and modern state-space model (SSM) architectures in the natural language domain. Similar to SSMs, xLSTMs have a linear runtime dependency on the sequence length and allow for constant-memory decoding at inference time, which makes them prime candidates for modeling long-range dependencies in biological and chemical sequences. In this work, we tailor xLSTM towards these domains and propose a suite of architectural variants called Bio-xLSTM. Extensive experiments in three large domains, genomics, proteins, and chemistry, were performed to assess xLSTM's ability to model biological and chemical sequences. The results show that models based on Bio-xLSTM a) can serve as proficient generative models for DNA, protein, and chemical sequences, b) learn rich representations for those modalities, and c) can perform in-context learning for proteins and small molecules.
VN-EGNN: E-Equivariant Graph Neural Networks with Virtual Nodes Enhance Protein Binding Site Identification
Apr 10, 2024



Being able to identify regions within or around proteins, to which ligands can potentially bind, is an essential step to develop new drugs. Binding site identification methods can now profit from the availability of large amounts of 3D structures in protein structure databases or from AlphaFold predictions. Current binding site identification methods heavily rely on graph neural networks (GNNs), usually designed to output E(3)-equivariant predictions. Such methods turned out to be very beneficial for physics-related tasks like binding energy or motion trajectory prediction. However, the performance of GNNs at binding site identification is still limited potentially due to the lack of dedicated nodes that model hidden geometric entities, such as binding pockets. In this work, we extend E(n)-Equivariant Graph Neural Networks (EGNNs) by adding virtual nodes and applying an extended message passing scheme. The virtual nodes in these graphs are dedicated quantities to learn representations of binding sites, which leads to improved predictive performance. In our experiments, we show that our proposed method VN-EGNN sets a new state-of-the-art at locating binding site centers on COACH420, HOLO4K and PDBbind2020.
GNN-VPA: A Variance-Preserving Aggregation Strategy for Graph Neural Networks
Mar 07, 2024

Graph neural networks (GNNs), and especially message-passing neural networks, excel in various domains such as physics, drug discovery, and molecular modeling. The expressivity of GNNs with respect to their ability to discriminate non-isomorphic graphs critically depends on the functions employed for message aggregation and graph-level readout. By applying signal propagation theory, we propose a variance-preserving aggregation function (VPA) that maintains expressivity, but yields improved forward and backward dynamics. Experiments demonstrate that VPA leads to increased predictive performance for popular GNN architectures as well as improved learning dynamics. Our results could pave the way towards normalizer-free or self-normalizing GNNs.
Redundancy-aware unsupervised rankings for collections of gene sets
Jul 30, 2023



The biological roles of gene sets are used to group them into collections. These collections are often characterized by being high-dimensional, overlapping, and redundant families of sets, thus precluding a straightforward interpretation and study of their content. Bioinformatics looked for solutions to reduce their dimension or increase their intepretability. One possibility lies in aggregating overlapping gene sets to create larger pathways, but the modified biological pathways are hardly biologically justifiable. We propose to use importance scores to rank the pathways in the collections studying the context from a set covering perspective. The proposed Shapley values-based scores consider the distribution of the singletons and the size of the sets in the families; Furthermore, a trick allows us to circumvent the usual exponential complexity of Shapley values' computation. Finally, we address the challenge of including a redundancy awareness in the obtained rankings where, in our case, sets are redundant if they show prominent intersections. The rankings can be used to reduce the dimension of collections of gene sets, such that they show lower redundancy and still a high coverage of the genes. We further investigate the impact of our selection on Gene Sets Enrichment Analysis. The proposed method shows a practical utility in bioinformatics to increase the interpretability of the collections of gene sets and a step forward to include redundancy into Shapley values computations.
Redundancy-aware unsupervised ranking based on game theory -- application to gene enrichment analysis
Jul 22, 2022
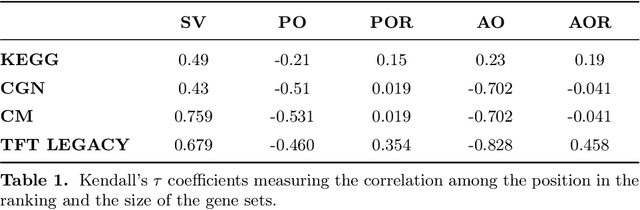

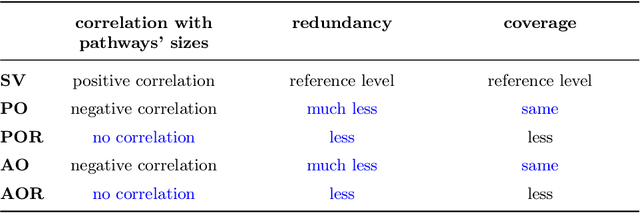
Gene set collections are a common ground to study the enrichment of genes for specific phenotypic traits. Gene set enrichment analysis aims to identify genes that are over-represented in gene sets collections and might be associated with a specific phenotypic trait. However, as this involves a massive number of hypothesis testing, it is often questionable whether a pre-processing step to reduce gene sets collections' sizes is helpful. Moreover, the often highly overlapping gene sets and the consequent low interpretability of gene sets' collections demand for a reduction of the included gene sets. Inspired by this bioinformatics context, we propose a method to rank sets within a family of sets based on the distribution of the singletons and their size. We obtain sets' importance scores by computing Shapley values without incurring into the usual exponential number of evaluations of the value function. Moreover, we address the challenge of including a redundancy awareness in the rankings obtained where, in our case, sets are redundant if they show prominent intersections. We finally evaluate our approach for gene sets collections; the rankings obtained show low redundancy and high coverage of the genes. The unsupervised nature of the proposed ranking does not allow for an evident increase in the number of significant gene sets for specific phenotypic traits when reducing the size of the collections. However, we believe that the rankings proposed are of use in bioinformatics to increase interpretability of the gene sets collections and a step forward to include redundancy into Shapley values computations.
Unsupervised Features Ranking via Coalitional Game Theory for Categorical Data
May 17, 2022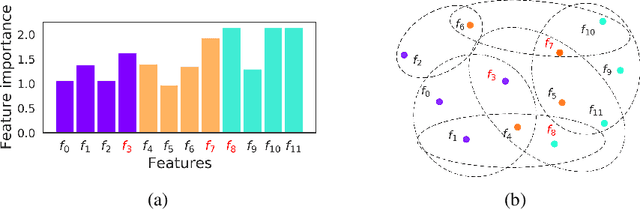
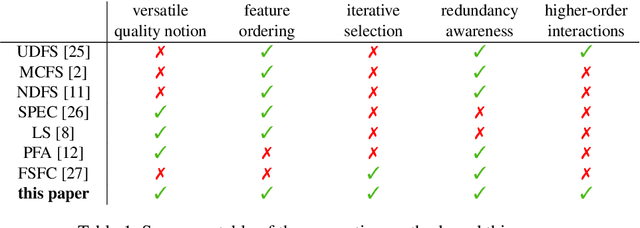
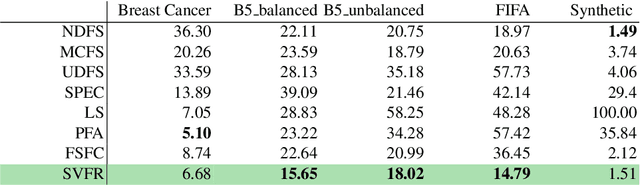
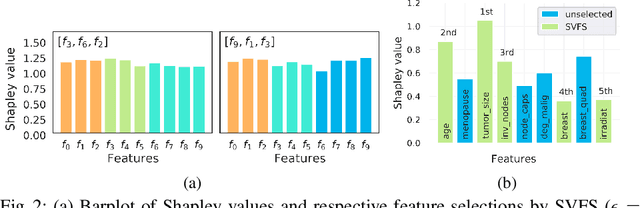
Not all real-world data are labeled, and when labels are not available, it is often costly to obtain them. Moreover, as many algorithms suffer from the curse of dimensionality, reducing the features in the data to a smaller set is often of great utility. Unsupervised feature selection aims to reduce the number of features, often using feature importance scores to quantify the relevancy of single features to the task at hand. These scores can be based only on the distribution of variables and the quantification of their interactions. The previous literature, mainly investigating anomaly detection and clusters, fails to address the redundancy-elimination issue. We propose an evaluation of correlations among features to compute feature importance scores representing the contribution of single features in explaining the dataset's structure. Based on Coalitional Game Theory, our feature importance scores include a notion of redundancy awareness making them a tool to achieve redundancy-free feature selection. We show that the deriving features' selection outperforms competing methods in lowering the redundancy rate while maximizing the information contained in the data. We also introduce an approximated version of the algorithm to reduce the complexity of Shapley values' computations.
Boundary Graph Neural Networks for 3D Simulations
Jun 21, 2021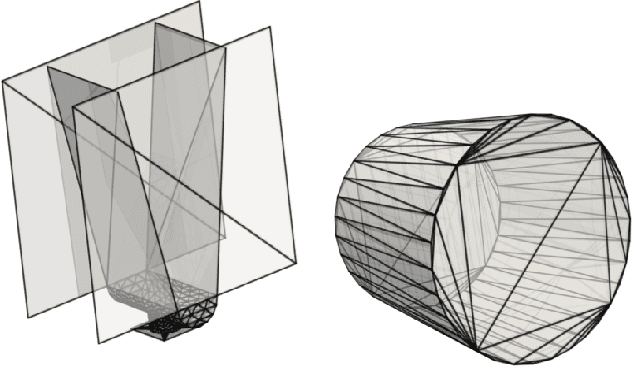

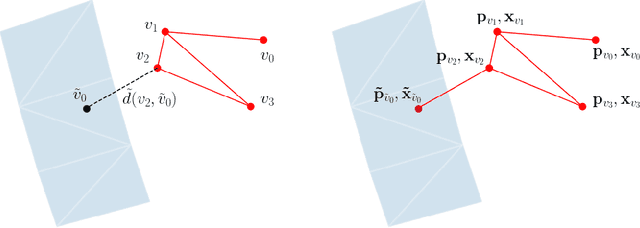
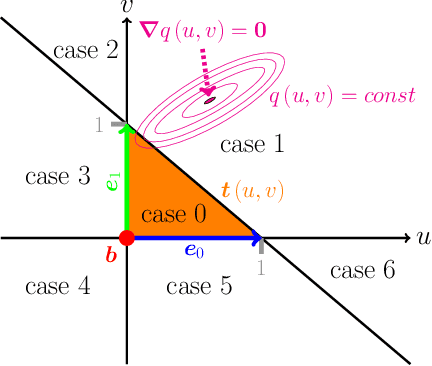
The abundance of data has given machine learning huge momentum in natural sciences and engineering. However, the modeling of simulated physical processes remains difficult. A key problem in doing so is the correct handling of geometric boundaries. While triangularized geometric boundaries are very common in engineering applications, they are notoriously difficult to model by machine learning approaches due to their heterogeneity with respect to size and orientation. In this work, we introduce Boundary Graph Neural Networks (BGNNs), which dynamically modify graph structures to address boundary conditions. Boundary graph structures are constructed via modifying edges, augmenting node features, and dynamically inserting virtual nodes. The new BGNNs are tested on complex 3D granular flow processes of hoppers and rotating drums which are standard parts of industrial machinery. Using precise simulations that are obtained by an expensive and complex discrete element method, BGNNs are evaluated in terms of computational efficiency as well as prediction accuracy of particle flows and mixing entropies. Even if complex boundaries are present, BGNNs are able to accurately reproduce 3D granular flows within simulation uncertainties over hundreds of thousands of simulation timesteps, and most notably particles completely stay within the geometric objects without using handcrafted conditions or restrictions.
Learning 3D Granular Flow Simulations
May 04, 2021


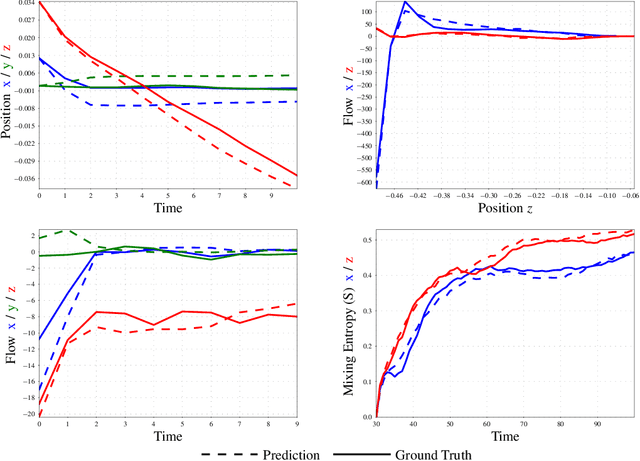
Recently, the application of machine learning models has gained momentum in natural sciences and engineering, which is a natural fit due to the abundance of data in these fields. However, the modeling of physical processes from simulation data without first principle solutions remains difficult. Here, we present a Graph Neural Networks approach towards accurate modeling of complex 3D granular flow simulation processes created by the discrete element method LIGGGHTS and concentrate on simulations of physical systems found in real world applications like rotating drums and hoppers. We discuss how to implement Graph Neural Networks that deal with 3D objects, boundary conditions, particle - particle, and particle - boundary interactions such that an accurate modeling of relevant physical quantities is made possible. Finally, we compare the machine learning based trajectories to LIGGGHTS trajectories in terms of particle flows and mixing entropies.