 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeOn the Consistency and Robustness of Saliency Explanations for Time Series Classification
Sep 04, 2023



Interpretable machine learning and explainable artificial intelligence have become essential in many applications. The trade-off between interpretability and model performance is the traitor to developing intrinsic and model-agnostic interpretation methods. Although model explanation approaches have achieved significant success in vision and natural language domains, explaining time series remains challenging. The complex pattern in the feature domain, coupled with the additional temporal dimension, hinders efficient interpretation. Saliency maps have been applied to interpret time series windows as images. However, they are not naturally designed for sequential data, thus suffering various issues. This paper extensively analyzes the consistency and robustness of saliency maps for time series features and temporal attribution. Specifically, we examine saliency explanations from both perturbation-based and gradient-based explanation models in a time series classification task. Our experimental results on five real-world datasets show that they all lack consistent and robust performances to some extent. By drawing attention to the flawed saliency explanation models, we motivate to develop consistent and robust explanations for time series classification.
Redundancy-aware unsupervised rankings for collections of gene sets
Jul 30, 2023



The biological roles of gene sets are used to group them into collections. These collections are often characterized by being high-dimensional, overlapping, and redundant families of sets, thus precluding a straightforward interpretation and study of their content. Bioinformatics looked for solutions to reduce their dimension or increase their intepretability. One possibility lies in aggregating overlapping gene sets to create larger pathways, but the modified biological pathways are hardly biologically justifiable. We propose to use importance scores to rank the pathways in the collections studying the context from a set covering perspective. The proposed Shapley values-based scores consider the distribution of the singletons and the size of the sets in the families; Furthermore, a trick allows us to circumvent the usual exponential complexity of Shapley values' computation. Finally, we address the challenge of including a redundancy awareness in the obtained rankings where, in our case, sets are redundant if they show prominent intersections. The rankings can be used to reduce the dimension of collections of gene sets, such that they show lower redundancy and still a high coverage of the genes. We further investigate the impact of our selection on Gene Sets Enrichment Analysis. The proposed method shows a practical utility in bioinformatics to increase the interpretability of the collections of gene sets and a step forward to include redundancy into Shapley values computations.
Redundancy-aware unsupervised ranking based on game theory -- application to gene enrichment analysis
Jul 22, 2022
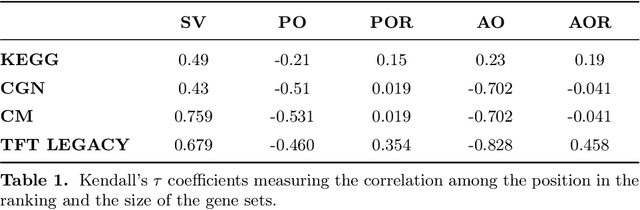

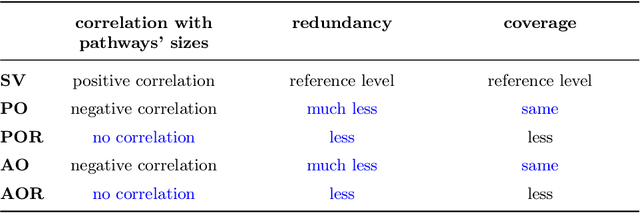
Gene set collections are a common ground to study the enrichment of genes for specific phenotypic traits. Gene set enrichment analysis aims to identify genes that are over-represented in gene sets collections and might be associated with a specific phenotypic trait. However, as this involves a massive number of hypothesis testing, it is often questionable whether a pre-processing step to reduce gene sets collections' sizes is helpful. Moreover, the often highly overlapping gene sets and the consequent low interpretability of gene sets' collections demand for a reduction of the included gene sets. Inspired by this bioinformatics context, we propose a method to rank sets within a family of sets based on the distribution of the singletons and their size. We obtain sets' importance scores by computing Shapley values without incurring into the usual exponential number of evaluations of the value function. Moreover, we address the challenge of including a redundancy awareness in the rankings obtained where, in our case, sets are redundant if they show prominent intersections. We finally evaluate our approach for gene sets collections; the rankings obtained show low redundancy and high coverage of the genes. The unsupervised nature of the proposed ranking does not allow for an evident increase in the number of significant gene sets for specific phenotypic traits when reducing the size of the collections. However, we believe that the rankings proposed are of use in bioinformatics to increase interpretability of the gene sets collections and a step forward to include redundancy into Shapley values computations.
Unsupervised Features Ranking via Coalitional Game Theory for Categorical Data
May 17, 2022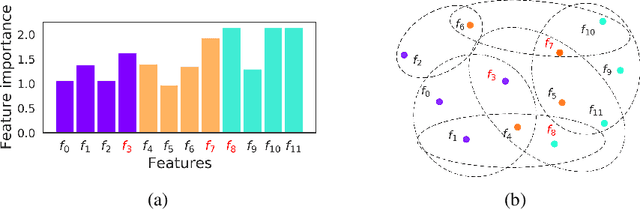
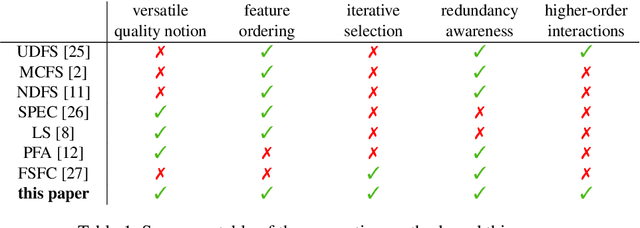
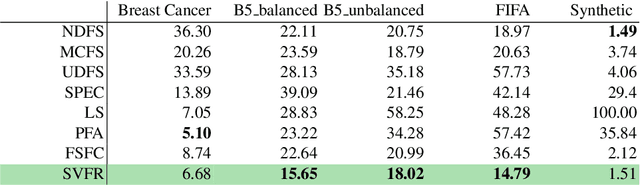
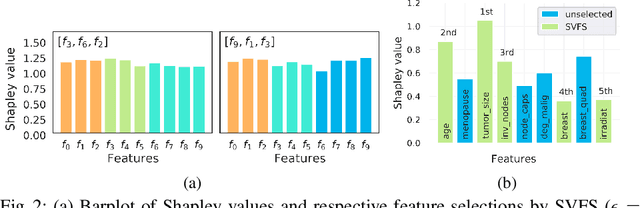
Not all real-world data are labeled, and when labels are not available, it is often costly to obtain them. Moreover, as many algorithms suffer from the curse of dimensionality, reducing the features in the data to a smaller set is often of great utility. Unsupervised feature selection aims to reduce the number of features, often using feature importance scores to quantify the relevancy of single features to the task at hand. These scores can be based only on the distribution of variables and the quantification of their interactions. The previous literature, mainly investigating anomaly detection and clusters, fails to address the redundancy-elimination issue. We propose an evaluation of correlations among features to compute feature importance scores representing the contribution of single features in explaining the dataset's structure. Based on Coalitional Game Theory, our feature importance scores include a notion of redundancy awareness making them a tool to achieve redundancy-free feature selection. We show that the deriving features' selection outperforms competing methods in lowering the redundancy rate while maximizing the information contained in the data. We also introduce an approximated version of the algorithm to reduce the complexity of Shapley values' computations.