 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeContrastive Geometric Learning Unlocks Unified Structure- and Ligand-Based Drug Design
Jan 14, 2026Structure-based and ligand-based computational drug design have traditionally relied on disjoint data sources and modeling assumptions, limiting their joint use at scale. In this work, we introduce Contrastive Geometric Learning for Unified Computational Drug Design (ConGLUDe), a single contrastive geometric model that unifies structure- and ligand-based training. ConGLUDe couples a geometric protein encoder that produces whole-protein representations and implicit embeddings of predicted binding sites with a fast ligand encoder, removing the need for pre-defined pockets. By aligning ligands with both global protein representations and multiple candidate binding sites through contrastive learning, ConGLUDe supports ligand-conditioned pocket prediction in addition to virtual screening and target fishing, while being trained jointly on protein-ligand complexes and large-scale bioactivity data. Across diverse benchmarks, ConGLUDe achieves state-of-the-art zero-shot virtual screening performance in settings where no binding pocket information is provided as input, substantially outperforms existing methods on a challenging target fishing task, and demonstrates competitive ligand-conditioned pocket selection. These results highlight the advantages of unified structure-ligand training and position ConGLUDe as a step toward general-purpose foundation models for drug discovery.
Bio-xLSTM: Generative modeling, representation and in-context learning of biological and chemical sequences
Nov 06, 2024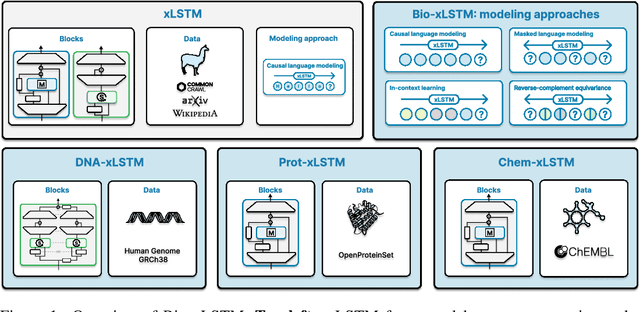
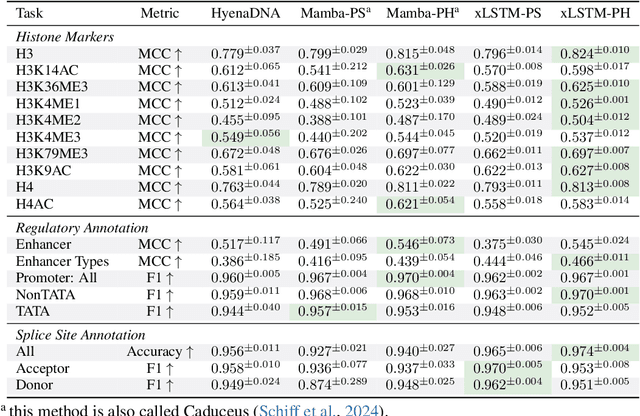
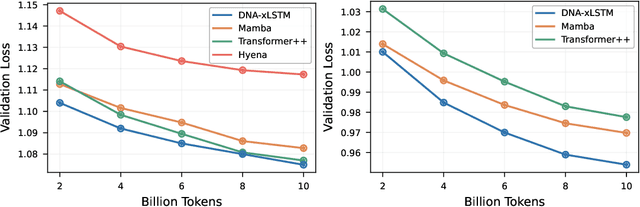
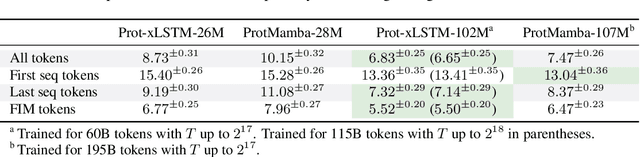
Language models for biological and chemical sequences enable crucial applications such as drug discovery, protein engineering, and precision medicine. Currently, these language models are predominantly based on Transformer architectures. While Transformers have yielded impressive results, their quadratic runtime dependency on the sequence length complicates their use for long genomic sequences and in-context learning on proteins and chemical sequences. Recently, the recurrent xLSTM architecture has been shown to perform favorably compared to Transformers and modern state-space model (SSM) architectures in the natural language domain. Similar to SSMs, xLSTMs have a linear runtime dependency on the sequence length and allow for constant-memory decoding at inference time, which makes them prime candidates for modeling long-range dependencies in biological and chemical sequences. In this work, we tailor xLSTM towards these domains and propose a suite of architectural variants called Bio-xLSTM. Extensive experiments in three large domains, genomics, proteins, and chemistry, were performed to assess xLSTM's ability to model biological and chemical sequences. The results show that models based on Bio-xLSTM a) can serve as proficient generative models for DNA, protein, and chemical sequences, b) learn rich representations for those modalities, and c) can perform in-context learning for proteins and small molecules.
VN-EGNN: E-Equivariant Graph Neural Networks with Virtual Nodes Enhance Protein Binding Site Identification
Apr 10, 2024



Being able to identify regions within or around proteins, to which ligands can potentially bind, is an essential step to develop new drugs. Binding site identification methods can now profit from the availability of large amounts of 3D structures in protein structure databases or from AlphaFold predictions. Current binding site identification methods heavily rely on graph neural networks (GNNs), usually designed to output E(3)-equivariant predictions. Such methods turned out to be very beneficial for physics-related tasks like binding energy or motion trajectory prediction. However, the performance of GNNs at binding site identification is still limited potentially due to the lack of dedicated nodes that model hidden geometric entities, such as binding pockets. In this work, we extend E(n)-Equivariant Graph Neural Networks (EGNNs) by adding virtual nodes and applying an extended message passing scheme. The virtual nodes in these graphs are dedicated quantities to learn representations of binding sites, which leads to improved predictive performance. In our experiments, we show that our proposed method VN-EGNN sets a new state-of-the-art at locating binding site centers on COACH420, HOLO4K and PDBbind2020.
GNN-VPA: A Variance-Preserving Aggregation Strategy for Graph Neural Networks
Mar 07, 2024

Graph neural networks (GNNs), and especially message-passing neural networks, excel in various domains such as physics, drug discovery, and molecular modeling. The expressivity of GNNs with respect to their ability to discriminate non-isomorphic graphs critically depends on the functions employed for message aggregation and graph-level readout. By applying signal propagation theory, we propose a variance-preserving aggregation function (VPA) that maintains expressivity, but yields improved forward and backward dynamics. Experiments demonstrate that VPA leads to increased predictive performance for popular GNN architectures as well as improved learning dynamics. Our results could pave the way towards normalizer-free or self-normalizing GNNs.