 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGradient Guided Hypotheses: A unified solution to enable machine learning models on scarce and noisy data regimes
May 29, 2024



Ensuring high-quality data is paramount for maximizing the performance of machine learning models and business intelligence systems. However, challenges in data quality, including noise in data capture, missing records, limited data production, and confounding variables, significantly constrain the potential performance of these systems. In this study, we propose an architecture-agnostic algorithm, Gradient Guided Hypotheses (GGH), designed to address these challenges. GGH analyses gradients from hypotheses as a proxy of distinct and possibly contradictory patterns in the data. This framework entails an additional step in machine learning training, where gradients can be included or excluded from backpropagation. In this manner, missing and noisy data are addressed through a unified solution that perceives both challenges as facets of the same overarching issue: the propagation of erroneous information. Experimental validation of GGH is conducted using real-world open-source datasets, where records with missing rates of up to 98.5% are simulated. Comparative analysis with state-of-the-art imputation methods demonstrates a substantial improvement in model performance achieved by GGH. Specifically in very high scarcity regimes, GGH was found to be the only viable solution. Additionally, GGH's noise detection capabilities are showcased by introducing simulated noise into the datasets and observing enhanced model performance after filtering out the noisy data. This study presents GGH as a promising solution for improving data quality and model performance in various applications.
Modern Hopfield Networks for Few- and Zero-Shot Reaction Prediction
Apr 07, 2021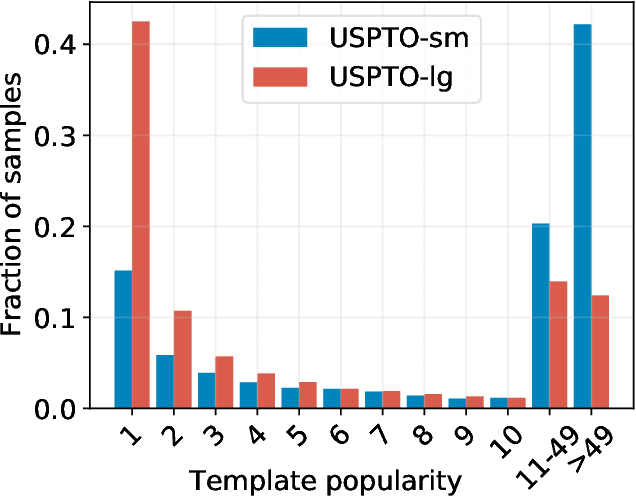
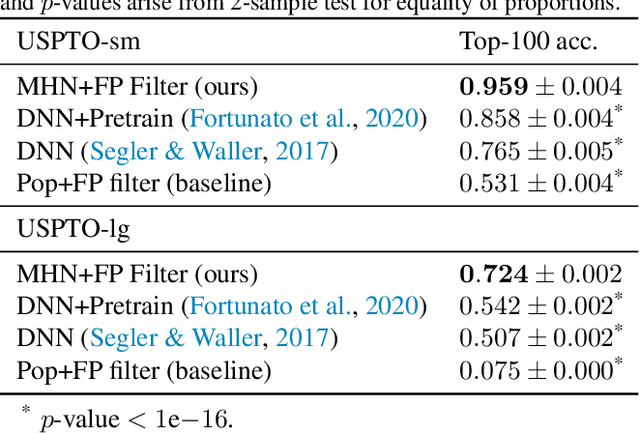
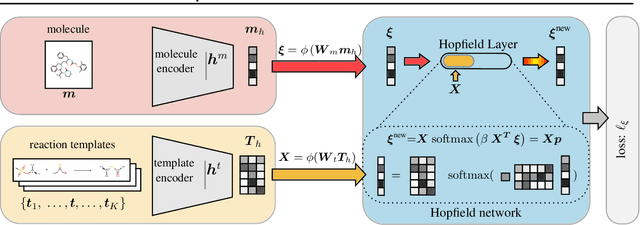
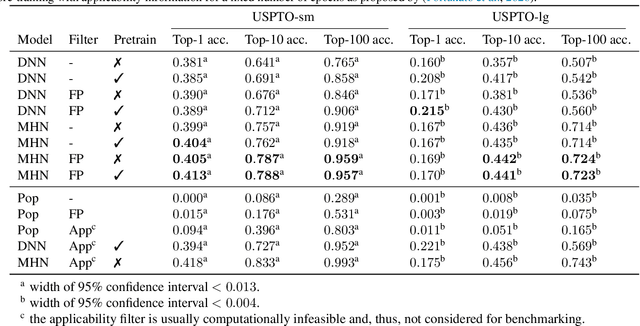
An essential step in the discovery of new drugs and materials is the synthesis of a molecule that exists so far only as an idea to test its biological and physical properties. While computer-aided design of virtual molecules has made large progress, computer-assisted synthesis planning (CASP) to realize physical molecules is still in its infancy and lacks a performance level that would enable large-scale molecule discovery. CASP supports the search for multi-step synthesis routes, which is very challenging due to high branching factors in each synthesis step and the hidden rules that govern the reactions. The central and repeatedly applied step in CASP is reaction prediction, for which machine learning methods yield the best performance. We propose a novel reaction prediction approach that uses a deep learning architecture with modern Hopfield networks (MHNs) that is optimized by contrastive learning. An MHN is an associative memory that can store and retrieve chemical reactions in each layer of a deep learning architecture. We show that our MHN contrastive learning approach enables few- and zero-shot learning for reaction prediction which, in contrast to previous methods, can deal with rare, single, or even no training example(s) for a reaction. On a well established benchmark, our MHN approach pushes the state-of-the-art performance up by a large margin as it improves the predictive top-100 accuracy from $0.858\pm0.004$ to $0.959\pm0.004$. This advance might pave the way to large-scale molecule discovery.