 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLow-count Time Series Anomaly Detection
Aug 24, 2023



Low-count time series describe sparse or intermittent events, which are prevalent in large-scale online platforms that capture and monitor diverse data types. Several distinct challenges surface when modelling low-count time series, particularly low signal-to-noise ratios (when anomaly signatures are provably undetectable), and non-uniform performance (when average metrics are not representative of local behaviour). The time series anomaly detection community currently lacks explicit tooling and processes to model and reliably detect anomalies in these settings. We address this gap by introducing a novel generative procedure for creating benchmark datasets comprising of low-count time series with anomalous segments. Via a mixture of theoretical and empirical analysis, our work explains how widely-used algorithms struggle with the distribution overlap between normal and anomalous segments. In order to mitigate this shortcoming, we then leverage our findings to demonstrate how anomaly score smoothing consistently improves performance. The practical utility of our analysis and recommendation is validated on a real-world dataset containing sales data for retail stores.
Understanding the Effects of Dataset Characteristics on Offline Reinforcement Learning
Nov 08, 2021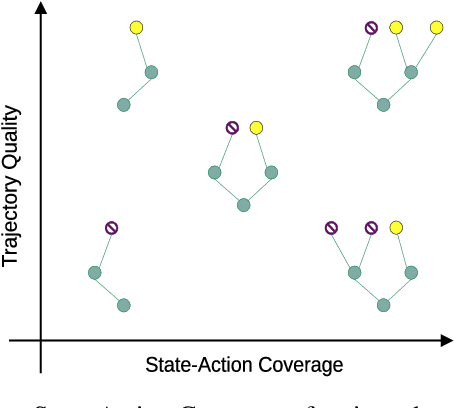
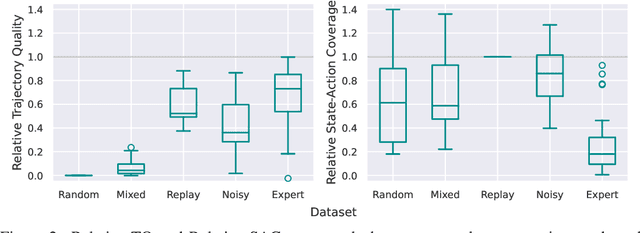
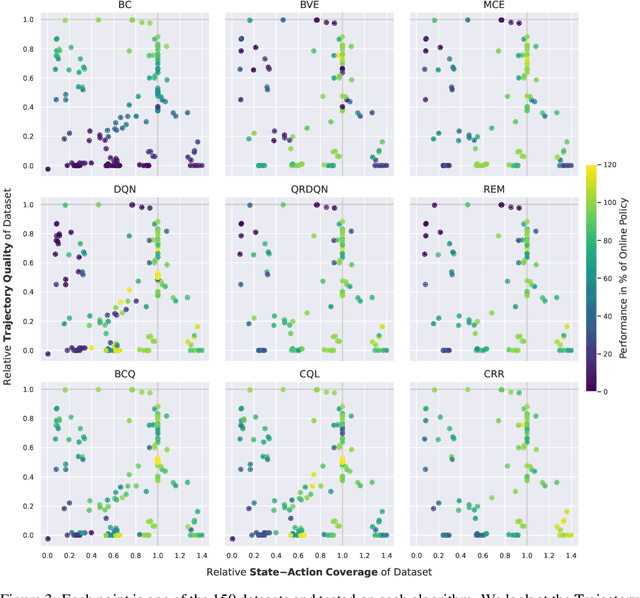

In real world, affecting the environment by a weak policy can be expensive or very risky, therefore hampers real world applications of reinforcement learning. Offline Reinforcement Learning (RL) can learn policies from a given dataset without interacting with the environment. However, the dataset is the only source of information for an Offline RL algorithm and determines the performance of the learned policy. We still lack studies on how dataset characteristics influence different Offline RL algorithms. Therefore, we conducted a comprehensive empirical analysis of how dataset characteristics effect the performance of Offline RL algorithms for discrete action environments. A dataset is characterized by two metrics: (1) the average dataset return measured by the Trajectory Quality (TQ) and (2) the coverage measured by the State-Action Coverage (SACo). We found that variants of the off-policy Deep Q-Network family require datasets with high SACo to perform well. Algorithms that constrain the learned policy towards the given dataset perform well for datasets with high TQ or SACo. For datasets with high TQ, Behavior Cloning outperforms or performs similarly to the best Offline RL algorithms.
Modern Hopfield Networks for Few- and Zero-Shot Reaction Prediction
Apr 07, 2021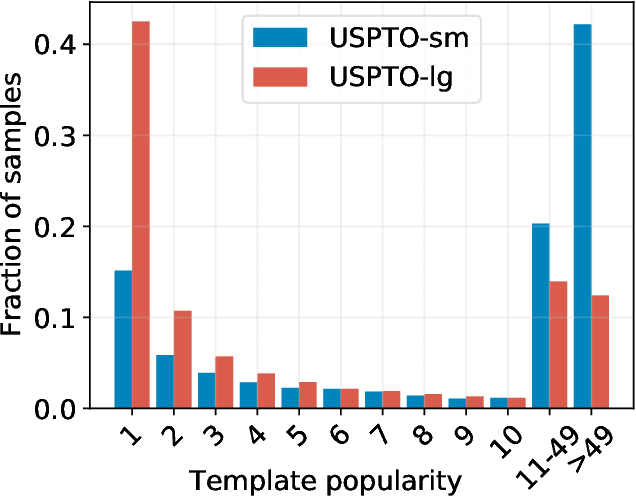
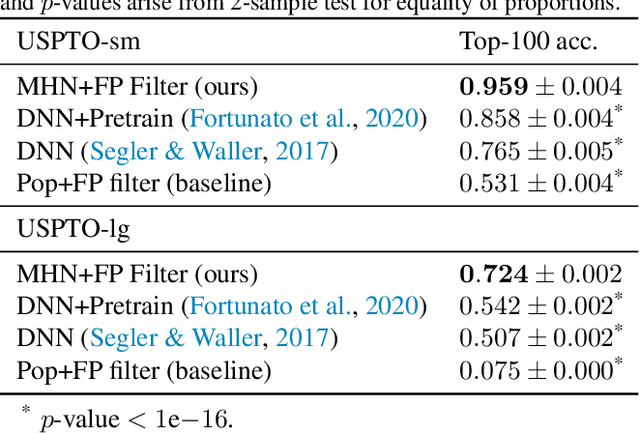
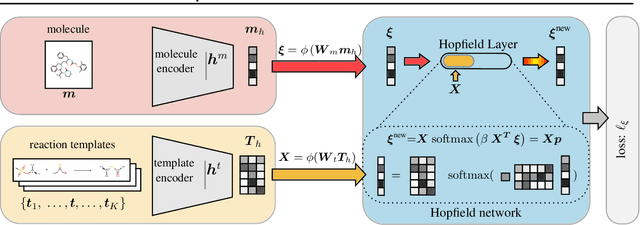
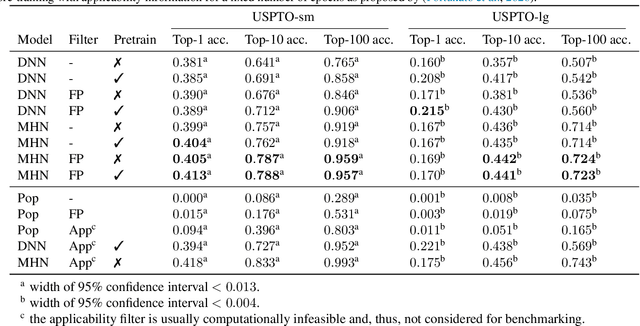
An essential step in the discovery of new drugs and materials is the synthesis of a molecule that exists so far only as an idea to test its biological and physical properties. While computer-aided design of virtual molecules has made large progress, computer-assisted synthesis planning (CASP) to realize physical molecules is still in its infancy and lacks a performance level that would enable large-scale molecule discovery. CASP supports the search for multi-step synthesis routes, which is very challenging due to high branching factors in each synthesis step and the hidden rules that govern the reactions. The central and repeatedly applied step in CASP is reaction prediction, for which machine learning methods yield the best performance. We propose a novel reaction prediction approach that uses a deep learning architecture with modern Hopfield networks (MHNs) that is optimized by contrastive learning. An MHN is an associative memory that can store and retrieve chemical reactions in each layer of a deep learning architecture. We show that our MHN contrastive learning approach enables few- and zero-shot learning for reaction prediction which, in contrast to previous methods, can deal with rare, single, or even no training example(s) for a reaction. On a well established benchmark, our MHN approach pushes the state-of-the-art performance up by a large margin as it improves the predictive top-100 accuracy from $0.858\pm0.004$ to $0.959\pm0.004$. This advance might pave the way to large-scale molecule discovery.
Large-scale ligand-based virtual screening for SARS-CoV-2 inhibitors using deep neural networks
Apr 03, 2020

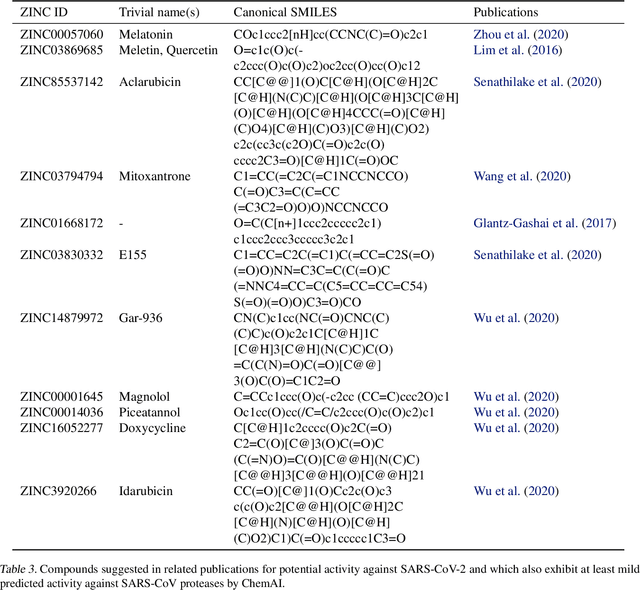
Due to the current severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic, there is an urgent need for novel therapies and drugs. We conducted a large-scale virtual screening for small molecules that are potential CoV-2 inhibitors. To this end, we utilized "ChemAI", a deep neural network trained on more than 220M data points across 3.6M molecules from three public drug-discovery databases. With ChemAI, we screened and ranked one billion molecules from the ZINC database for favourable effects against CoV-2. We then reduced the result to the 30,000 top-ranked compounds, which are readily accessible and purchasable via the ZINC database. Additionally, we screened the DrugBank using ChemAI to allow for drug repurposing, which would be a fast way towards a therapy. We provide these top-ranked compounds of ZINC and DrugBank as a library for further screening with bioassays at https://github.com/ml-jku/sars-cov-inhibitors-chemai.
Fréchet ChemNet Distance: A metric for generative models for molecules in drug discovery
Aug 01, 2018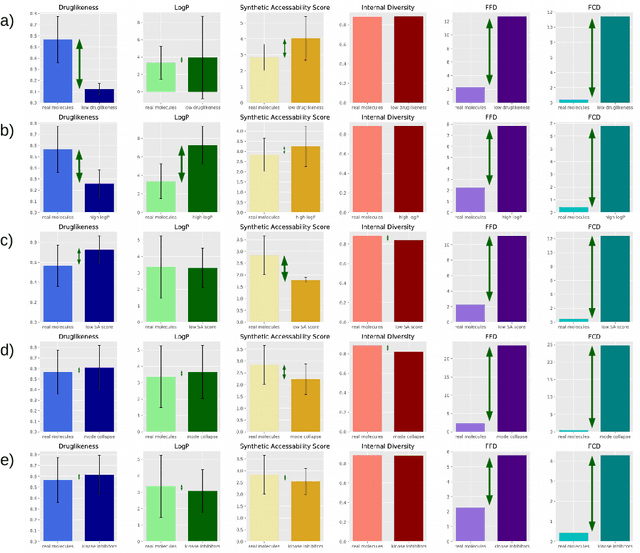
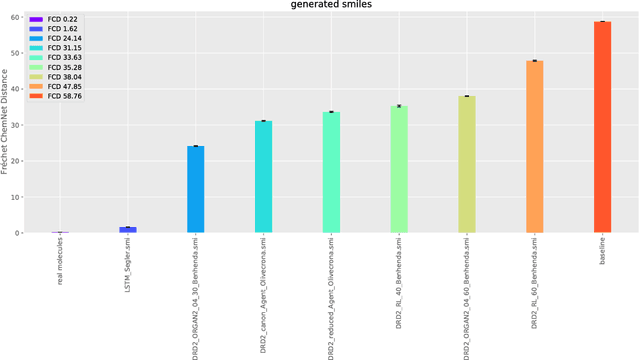
The new wave of successful generative models in machine learning has increased the interest in deep learning driven de novo drug design. However, assessing the performance of such generative models is notoriously difficult. Metrics that are typically used to assess the performance of such generative models are the percentage of chemically valid molecules or the similarity to real molecules in terms of particular descriptors, such as the partition coefficient (logP) or druglikeness. However, method comparison is difficult because of the inconsistent use of evaluation metrics, the necessity for multiple metrics, and the fact that some of these measures can easily be tricked by simple rule-based systems. We propose a novel distance measure between two sets of molecules, called Fr\'echet ChemNet distance (FCD), that can be used as an evaluation metric for generative models. The FCD is similar to a recently established performance metric for comparing image generation methods, the Fr\'echet Inception Distance (FID). Whereas the FID uses one of the hidden layers of InceptionNet, the FCD utilizes the penultimate layer of a deep neural network called ChemNet, which was trained to predict drug activities. Thus, the FCD metric takes into account chemically and biologically relevant information about molecules, and also measures the diversity of the set via the distribution of generated molecules. The FCD's advantage over previous metrics is that it can detect if generated molecules are a) diverse and have similar b) chemical and c) biological properties as real molecules. We further provide an easy-to-use implementation that only requires the SMILES representation of the generated molecules as input to calculate the FCD. Implementations are available at: https://www.github.com/bioinf-jku/FCD