 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeInterpretable Deep Learning in Drug Discovery
Mar 18, 2019

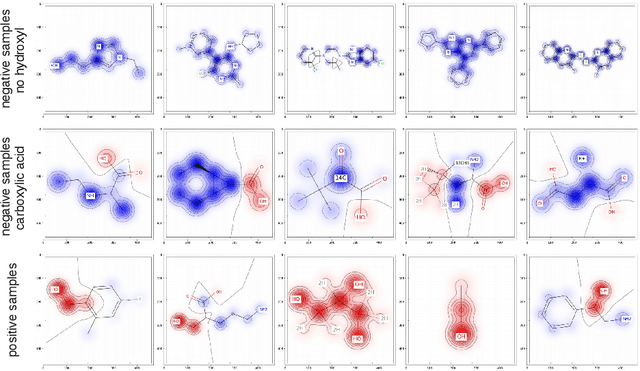
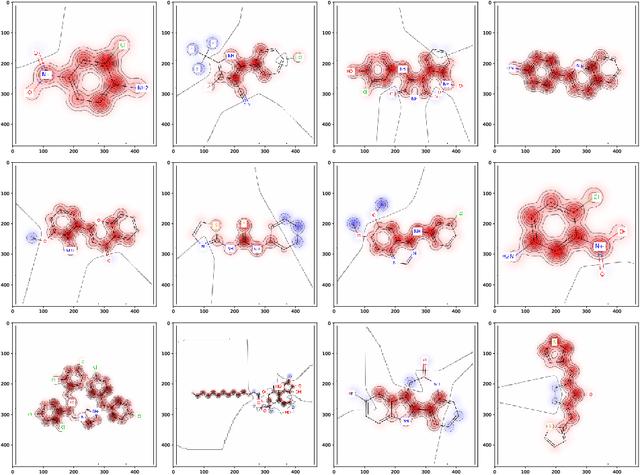
Without any means of interpretation, neural networks that predict molecular properties and bioactivities are merely black boxes. We will unravel these black boxes and will demonstrate approaches to understand the learned representations which are hidden inside these models. We show how single neurons can be interpreted as classifiers which determine the presence or absence of pharmacophore- or toxicophore-like structures, thereby generating new insights and relevant knowledge for chemistry, pharmacology and biochemistry. We further discuss how these novel pharmacophores/toxicophores can be determined from the network by identifying the most relevant components of a compound for the prediction of the network. Additionally, we propose a method which can be used to extract new pharmacophores from a model and will show that these extracted structures are consistent with literature findings. We envision that having access to such interpretable knowledge is a crucial aid in the development and design of new pharmaceutically active molecules, and helps to investigate and understand failures and successes of current methods.
Fréchet ChemNet Distance: A metric for generative models for molecules in drug discovery
Aug 01, 2018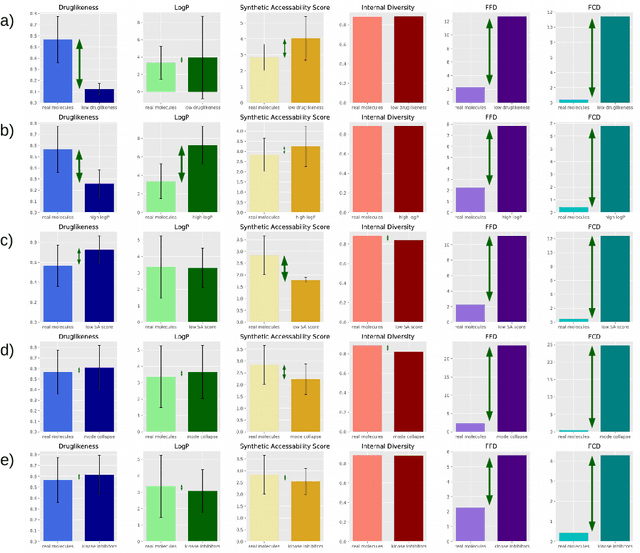
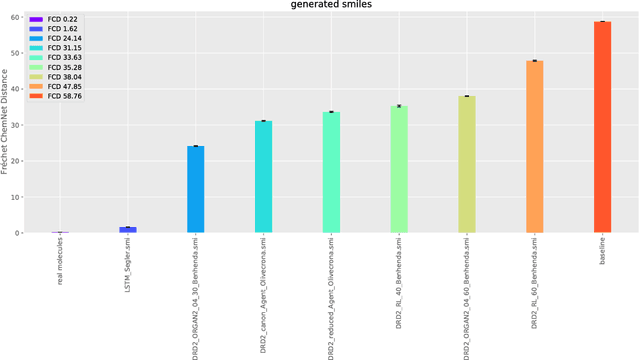
The new wave of successful generative models in machine learning has increased the interest in deep learning driven de novo drug design. However, assessing the performance of such generative models is notoriously difficult. Metrics that are typically used to assess the performance of such generative models are the percentage of chemically valid molecules or the similarity to real molecules in terms of particular descriptors, such as the partition coefficient (logP) or druglikeness. However, method comparison is difficult because of the inconsistent use of evaluation metrics, the necessity for multiple metrics, and the fact that some of these measures can easily be tricked by simple rule-based systems. We propose a novel distance measure between two sets of molecules, called Fr\'echet ChemNet distance (FCD), that can be used as an evaluation metric for generative models. The FCD is similar to a recently established performance metric for comparing image generation methods, the Fr\'echet Inception Distance (FID). Whereas the FID uses one of the hidden layers of InceptionNet, the FCD utilizes the penultimate layer of a deep neural network called ChemNet, which was trained to predict drug activities. Thus, the FCD metric takes into account chemically and biologically relevant information about molecules, and also measures the diversity of the set via the distribution of generated molecules. The FCD's advantage over previous metrics is that it can detect if generated molecules are a) diverse and have similar b) chemical and c) biological properties as real molecules. We further provide an easy-to-use implementation that only requires the SMILES representation of the generated molecules as input to calculate the FCD. Implementations are available at: https://www.github.com/bioinf-jku/FCD