 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeInterpretable Deep Learning in Drug Discovery
Paper and Code


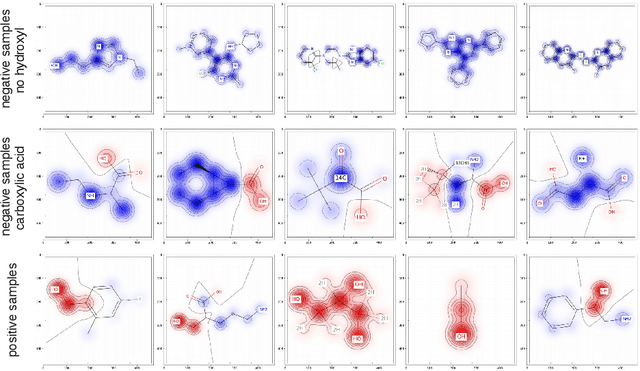
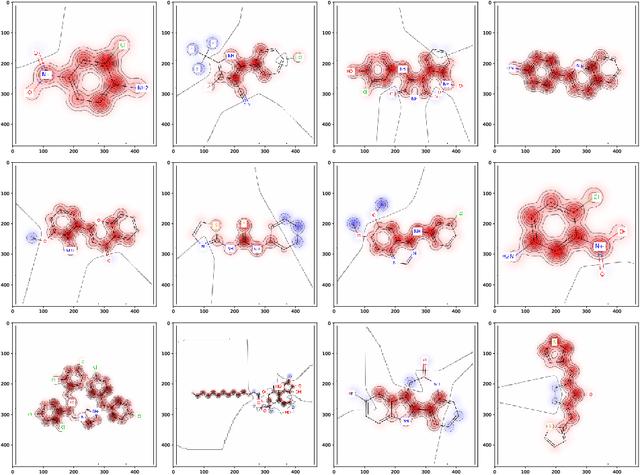
Without any means of interpretation, neural networks that predict molecular properties and bioactivities are merely black boxes. We will unravel these black boxes and will demonstrate approaches to understand the learned representations which are hidden inside these models. We show how single neurons can be interpreted as classifiers which determine the presence or absence of pharmacophore- or toxicophore-like structures, thereby generating new insights and relevant knowledge for chemistry, pharmacology and biochemistry. We further discuss how these novel pharmacophores/toxicophores can be determined from the network by identifying the most relevant components of a compound for the prediction of the network. Additionally, we propose a method which can be used to extract new pharmacophores from a model and will show that these extracted structures are consistent with literature findings. We envision that having access to such interpretable knowledge is a crucial aid in the development and design of new pharmaceutically active molecules, and helps to investigate and understand failures and successes of current methods.