 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCAVACHON: a hierarchical variational autoencoder to integrate multi-modal single-cell data
May 28, 2024



Paired single-cell sequencing technologies enable the simultaneous measurement of complementary modalities of molecular data at single-cell resolution. Along with the advances in these technologies, many methods based on variational autoencoders have been developed to integrate these data. However, these methods do not explicitly incorporate prior biological relationships between the data modalities, which could significantly enhance modeling and interpretation. We propose a novel probabilistic learning framework that explicitly incorporates conditional independence relationships between multi-modal data as a directed acyclic graph using a generalized hierarchical variational autoencoder. We demonstrate the versatility of our framework across various applications pertinent to single-cell multi-omics data integration. These include the isolation of common and distinct information from different modalities, modality-specific differential analysis, and integrated cell clustering. We anticipate that the proposed framework can facilitate the construction of highly flexible graphical models that can capture the complexities of biological hypotheses and unravel the connections between different biological data types, such as different modalities of paired single-cell multi-omics data. The implementation of the proposed framework can be found in the repository https://github.com/kuijjerlab/CAVACHON.
ImmunoLingo: Linguistics-based formalization of the antibody language
Sep 26, 2022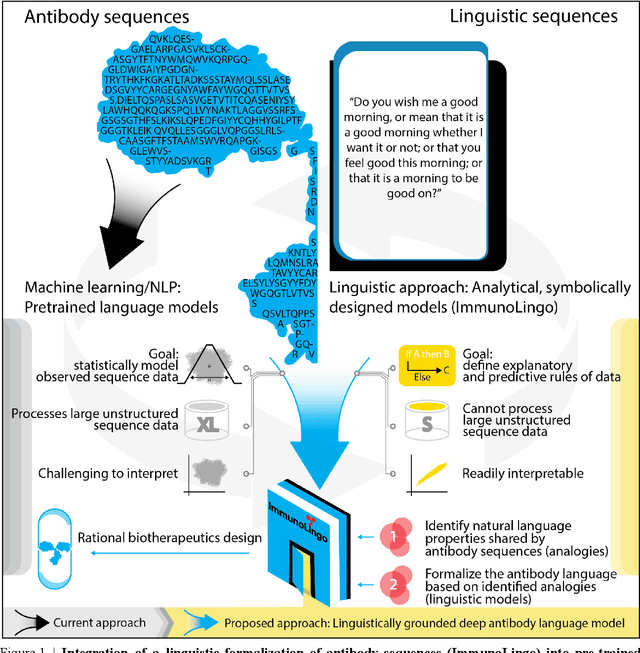
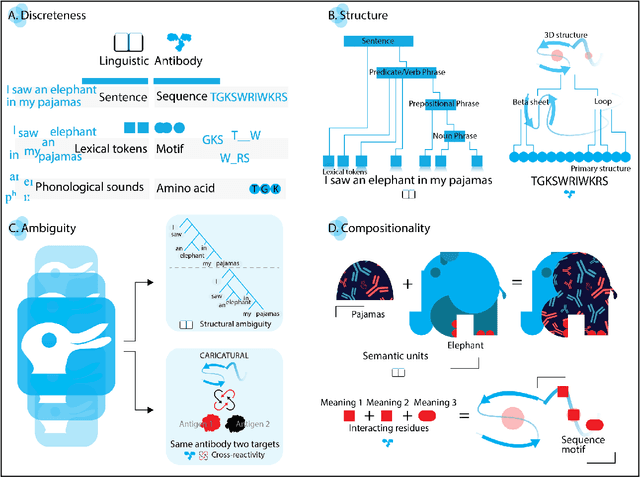
Apparent parallels between natural language and biological sequence have led to a recent surge in the application of deep language models (LMs) to the analysis of antibody and other biological sequences. However, a lack of a rigorous linguistic formalization of biological sequence languages, which would define basic components, such as lexicon (i.e., the discrete units of the language) and grammar (i.e., the rules that link sequence well-formedness, structure, and meaning) has led to largely domain-unspecific applications of LMs, which do not take into account the underlying structure of the biological sequences studied. A linguistic formalization, on the other hand, establishes linguistically-informed and thus domain-adapted components for LM applications. It would facilitate a better understanding of how differences and similarities between natural language and biological sequences influence the quality of LMs, which is crucial for the design of interpretable models with extractable sequence-functions relationship rules, such as the ones underlying the antibody specificity prediction problem. Deciphering the rules of antibody specificity is crucial to accelerating rational and in silico biotherapeutic drug design. Here, we formalize the properties of the antibody language and thereby establish not only a foundation for the application of linguistic tools in adaptive immune receptor analysis but also for the systematic immunolinguistic studies of immune receptor specificity in general.
Advancing protein language models with linguistics: a roadmap for improved interpretability
Jul 03, 2022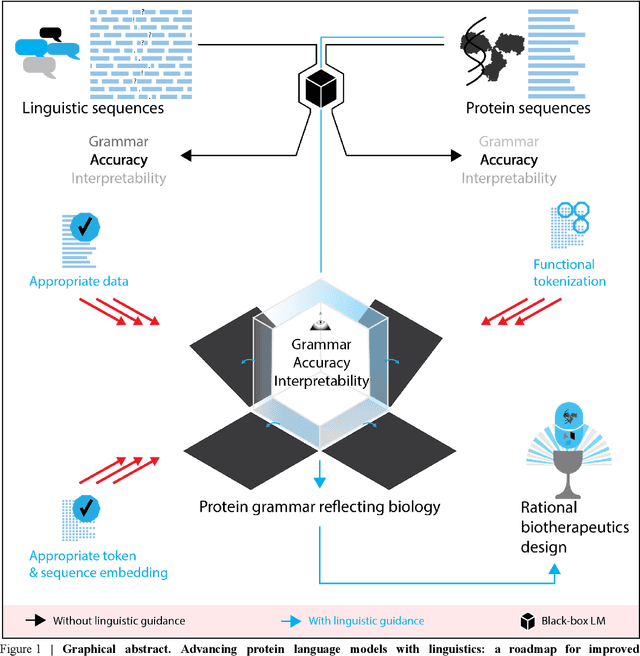
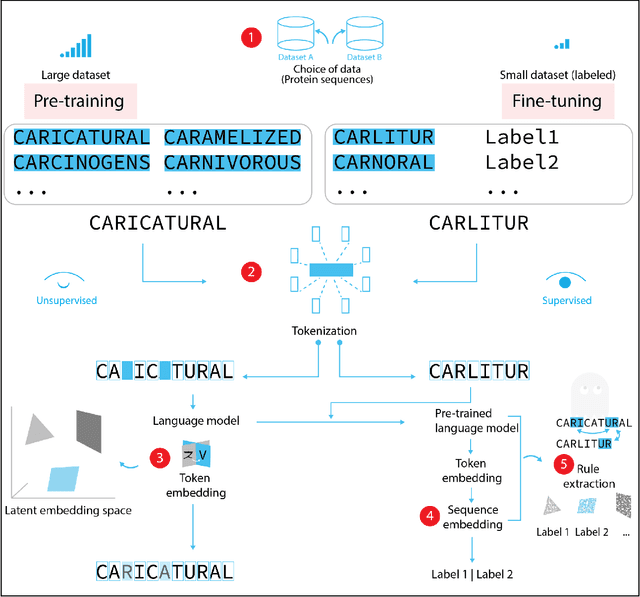
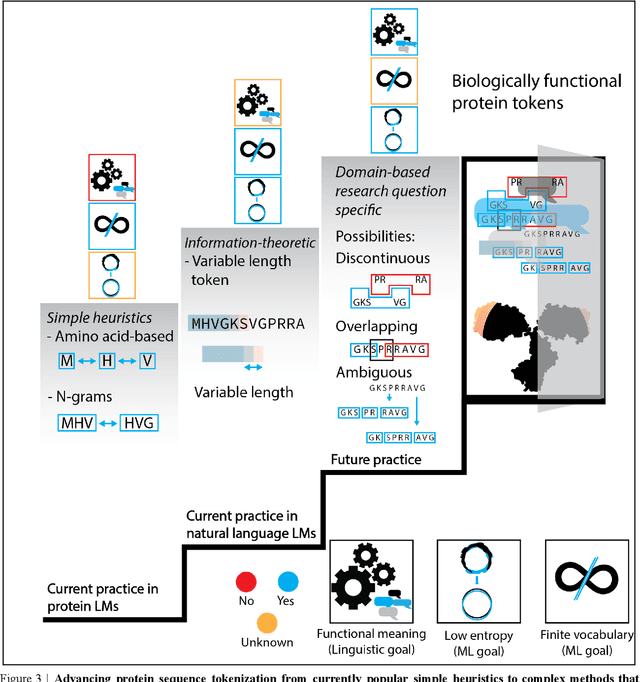
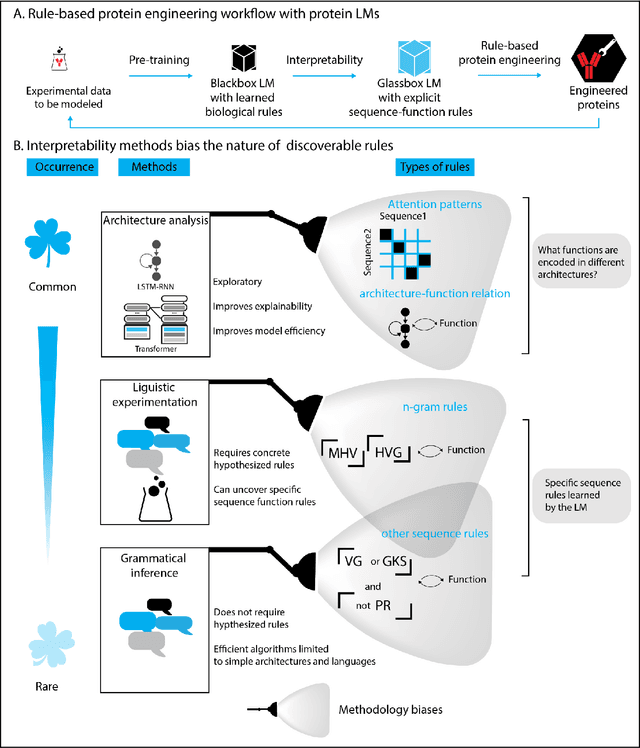
Deep neural-network-based language models (LMs) are increasingly applied to large-scale protein sequence data to predict protein function. However, being largely blackbox models and thus challenging to interpret, current protein LM approaches do not contribute to a fundamental understanding of sequence-function mappings, hindering rule-based biotherapeutic drug development. We argue that guidance drawn from linguistics, a field specialized in analytical rule extraction from natural language data, can aid with building more interpretable protein LMs that have learned relevant domain-specific rules. Differences between protein sequence data and linguistic sequence data require the integration of more domain-specific knowledge in protein LMs compared to natural language LMs. Here, we provide a linguistics-based roadmap for protein LM pipeline choices with regard to training data, tokenization, token embedding, sequence embedding, and model interpretation. Combining linguistics with protein LMs enables the development of next-generation interpretable machine learning models with the potential of uncovering the biological mechanisms underlying sequence-function relationships.
Improving generalization of machine learning-identified biomarkers with causal modeling: an investigation into immune receptor diagnostics
Apr 20, 2022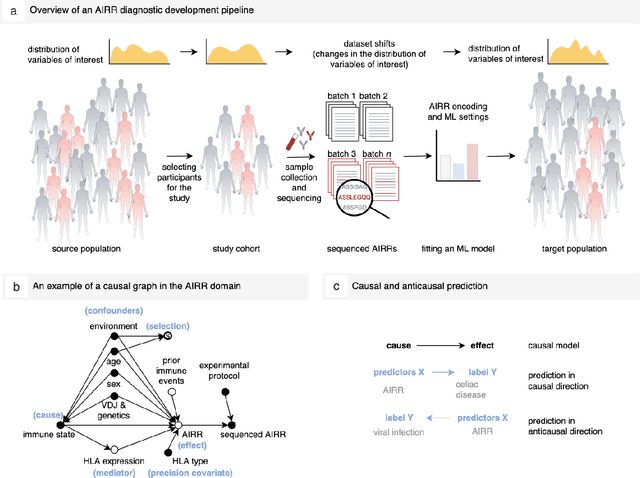
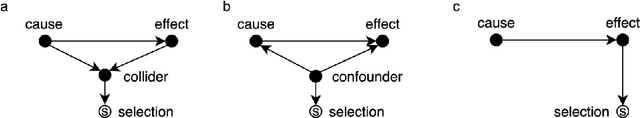
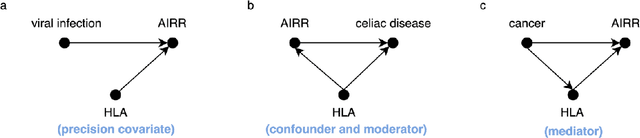
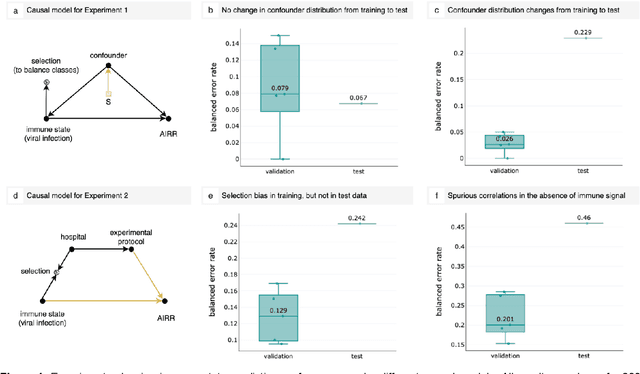
Machine learning is increasingly used to discover diagnostic and prognostic biomarkers from high-dimensional molecular data. However, a variety of factors related to experimental design may affect the ability to learn generalizable and clinically applicable diagnostics. Here, we argue that a causal perspective improves the identification of these challenges, and formalizes their relation to the robustness and generalization of machine learning-based diagnostics. To make for a concrete discussion, we focus on a specific, recently established high-dimensional biomarker - adaptive immune receptor repertoires (AIRRs). We discuss how the main biological and experimental factors of the AIRR domain may influence the learned biomarkers and provide easily adjustable simulations of such effects. In conclusion, we find that causal modeling improves machine learning-based biomarker robustness by identifying stable relations between variables and by guiding the adjustment of the relations and variables that vary between populations.
Modern Hopfield Networks and Attention for Immune Repertoire Classification
Jul 16, 2020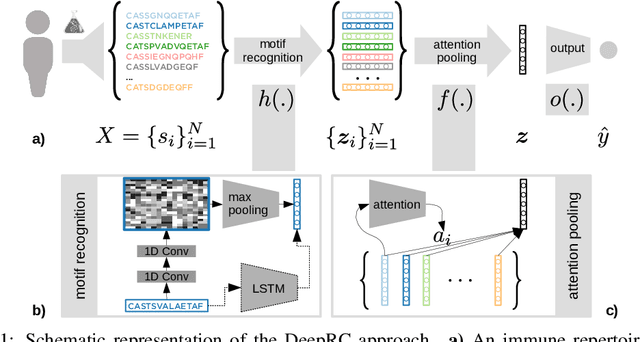
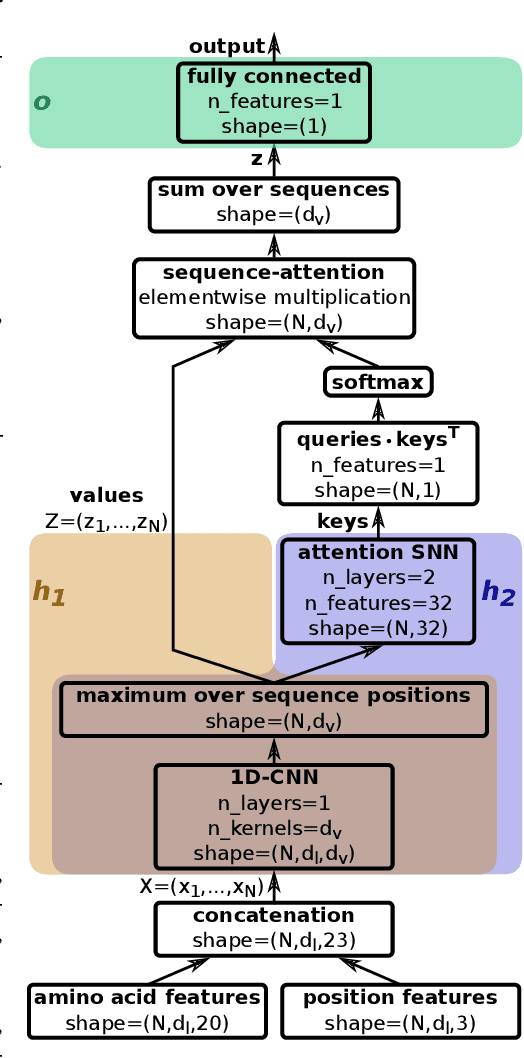
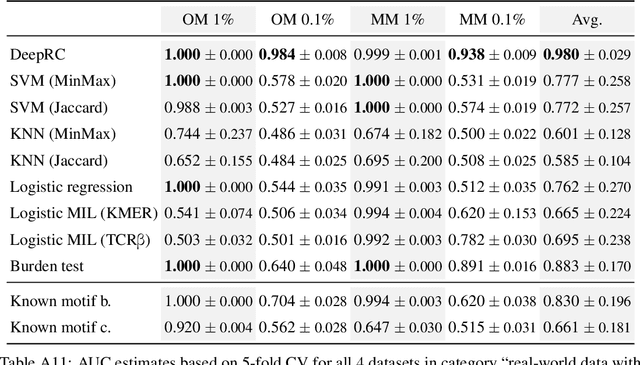
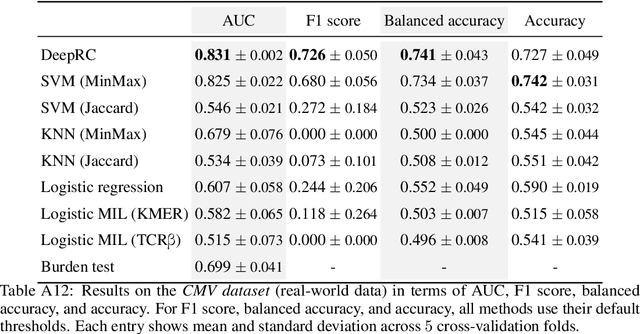
A central mechanism in machine learning is to identify, store, and recognize patterns. How to learn, access, and retrieve such patterns is crucial in Hopfield networks and the more recent transformer architectures. We show that the attention mechanism of transformer architectures is actually the update rule of modern Hopfield networks that can store exponentially many patterns. We exploit this high storage capacity of modern Hopfield networks to solve a challenging multiple instance learning (MIL) problem in computational biology: immune repertoire classification. Accurate and interpretable machine learning methods solving this problem could pave the way towards new vaccines and therapies, which is currently a very relevant research topic intensified by the COVID-19 crisis. Immune repertoire classification based on the vast number of immunosequences of an individual is a MIL problem with an unprecedentedly massive number of instances, two orders of magnitude larger than currently considered problems, and with an extremely low witness rate. In this work, we present our novel method DeepRC that integrates transformer-like attention, or equivalently modern Hopfield networks, into deep learning architectures for massive MIL such as immune repertoire classification. We demonstrate that DeepRC outperforms all other methods with respect to predictive performance on large-scale experiments, including simulated and real-world virus infection data, and enables the extraction of sequence motifs that are connected to a given disease class. Source code and datasets: https://github.com/ml-jku/DeepRC
Hopfield Networks is All You Need
Jul 16, 2020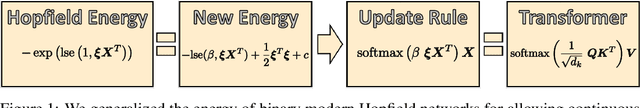
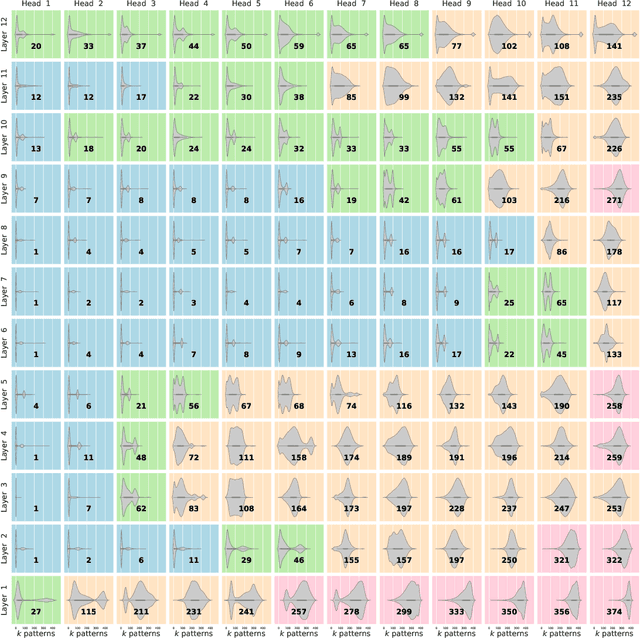
We show that the transformer attention mechanism is the update rule of a modern Hopfield network with continuous states. This new Hopfield network can store exponentially (with the dimension) many patterns, converges with one update, and has exponentially small retrieval errors. The number of stored patterns is traded off against convergence speed and retrieval error. The new Hopfield network has three types of energy minima (fixed points of the update): (1) global fixed point averaging over all patterns, (2) metastable states averaging over a subset of patterns, and (3) fixed points which store a single pattern. Transformer and BERT models operate in their first layers preferably in the global averaging regime, while they operate in higher layers in metastable states. The gradient in transformers is maximal for metastable states, is uniformly distributed for global averaging, and vanishes for a fixed point near a stored pattern. Using the Hopfield network interpretation, we analyzed learning of transformer and BERT models. Learning starts with attention heads that average and then most of them switch to metastable states. However, the majority of heads in the first layers still averages and can be replaced by averaging, e.g. our proposed Gaussian weighting. In contrast, heads in the last layers steadily learn and seem to use metastable states to collect information created in lower layers. These heads seem to be a promising target for improving transformers. Neural networks with Hopfield networks outperform other methods on immune repertoire classification, where the Hopfield net stores several hundreds of thousands of patterns. We provide a new PyTorch layer called "Hopfield", which allows to equip deep learning architectures with modern Hopfield networks as a new powerful concept comprising pooling, memory, and attention. GitHub: https://github.com/ml-jku/hopfield-layers