 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLearning to Plan Chemical Syntheses
Aug 14, 2017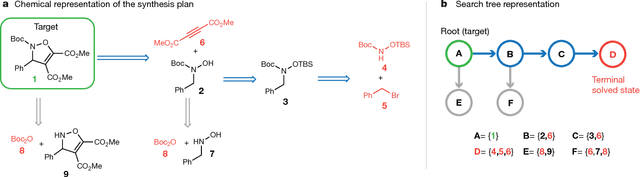
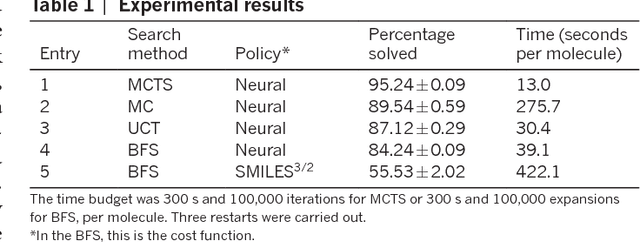
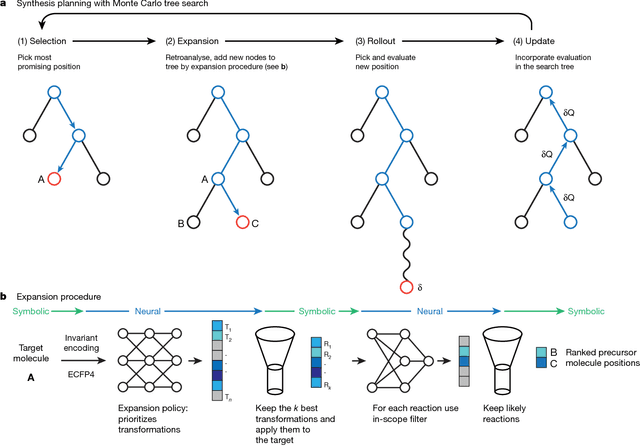
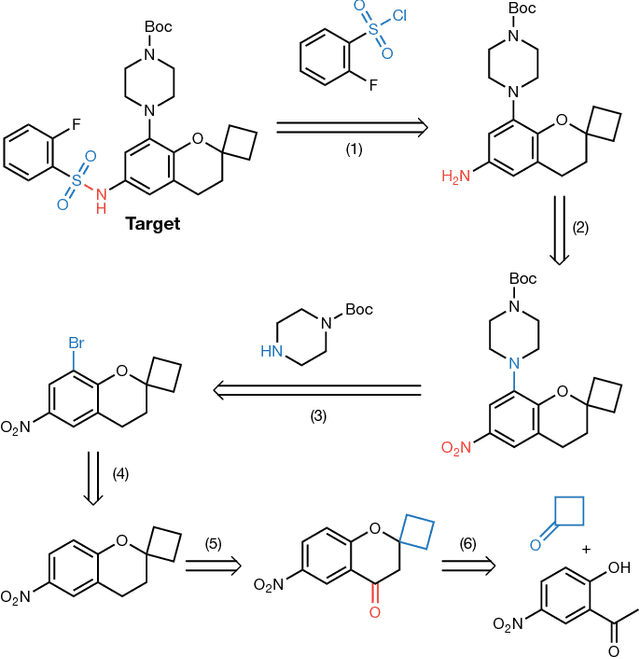
From medicines to materials, small organic molecules are indispensable for human well-being. To plan their syntheses, chemists employ a problem solving technique called retrosynthesis. In retrosynthesis, target molecules are recursively transformed into increasingly simpler precursor compounds until a set of readily available starting materials is obtained. Computer-aided retrosynthesis would be a highly valuable tool, however, past approaches were slow and provided results of unsatisfactory quality. Here, we employ Monte Carlo Tree Search (MCTS) to efficiently discover retrosynthetic routes. MCTS was combined with an expansion policy network that guides the search, and an "in-scope" filter network to pre-select the most promising retrosynthetic steps. These deep neural networks were trained on 12 million reactions, which represents essentially all reactions ever published in organic chemistry. Our system solves almost twice as many molecules and is 30 times faster in comparison to the traditional search method based on extracted rules and hand-coded heuristics. Finally after a 60 year history of computer-aided synthesis planning, chemists can no longer distinguish between routes generated by a computer system and real routes taken from the scientific literature. We anticipate that our method will accelerate drug and materials discovery by assisting chemists to plan better syntheses faster, and by enabling fully automated robot synthesis.
Towards "AlphaChem": Chemical Synthesis Planning with Tree Search and Deep Neural Network Policies
Jan 31, 2017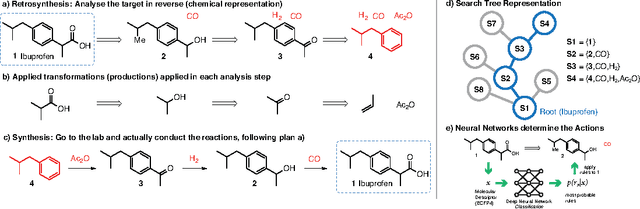
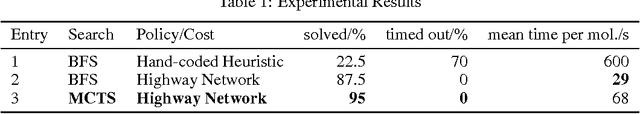
Retrosynthesis is a technique to plan the chemical synthesis of organic molecules, for example drugs, agro- and fine chemicals. In retrosynthesis, a search tree is built by analysing molecules recursively and dissecting them into simpler molecular building blocks until one obtains a set of known building blocks. The search space is intractably large, and it is difficult to determine the value of retrosynthetic positions. Here, we propose to model retrosynthesis as a Markov Decision Process. In combination with a Deep Neural Network policy learned from essentially the complete published knowledge of chemistry, Monte Carlo Tree Search (MCTS) can be used to evaluate positions. In exploratory studies, we demonstrate that MCTS with neural network policies outperforms the traditionally used best-first search with hand-coded heuristics.
Generating Focussed Molecule Libraries for Drug Discovery with Recurrent Neural Networks
Jan 05, 2017

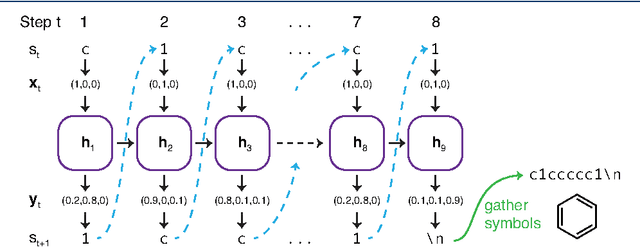
In de novo drug design, computational strategies are used to generate novel molecules with good affinity to the desired biological target. In this work, we show that recurrent neural networks can be trained as generative models for molecular structures, similar to statistical language models in natural language processing. We demonstrate that the properties of the generated molecules correlate very well with the properties of the molecules used to train the model. In order to enrich libraries with molecules active towards a given biological target, we propose to fine-tune the model with small sets of molecules, which are known to be active against that target. Against Staphylococcus aureus, the model reproduced 14% of 6051 hold-out test molecules that medicinal chemists designed, whereas against Plasmodium falciparum (Malaria) it reproduced 28% of 1240 test molecules. When coupled with a scoring function, our model can perform the complete de novo drug design cycle to generate large sets of novel molecules for drug discovery.
Modelling Chemical Reasoning to Predict Reactions
Aug 25, 2016


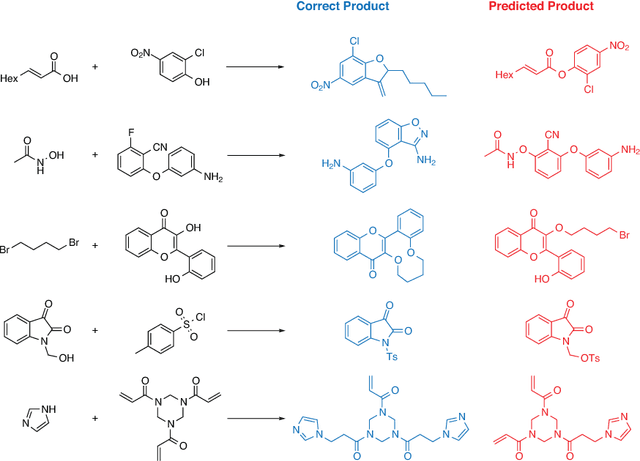
The ability to reason beyond established knowledge allows Organic Chemists to solve synthetic problems and to invent novel transformations. Here, we propose a model which mimics chemical reasoning and formalises reaction prediction as finding missing links in a knowledge graph. We have constructed a knowledge graph containing 14.4 million molecules and 8.2 million binary reactions, which represents the bulk of all chemical reactions ever published in the scientific literature. Our model outperforms a rule-based expert system in the reaction prediction task for 180,000 randomly selected binary reactions. We show that our data-driven model generalises even beyond known reaction types, and is thus capable of effectively (re-) discovering novel transformations (even including transition-metal catalysed reactions). Our model enables computers to infer hypotheses about reactivity and reactions by only considering the intrinsic local structure of the graph, and because each single reaction prediction is typically achieved in a sub-second time frame, our model can be used as a high-throughput generator of reaction hypotheses for reaction discovery.
* 17 pages, 8 figures