 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAccelerating high-throughput virtual screening through molecular pool-based active learning
Dec 13, 2020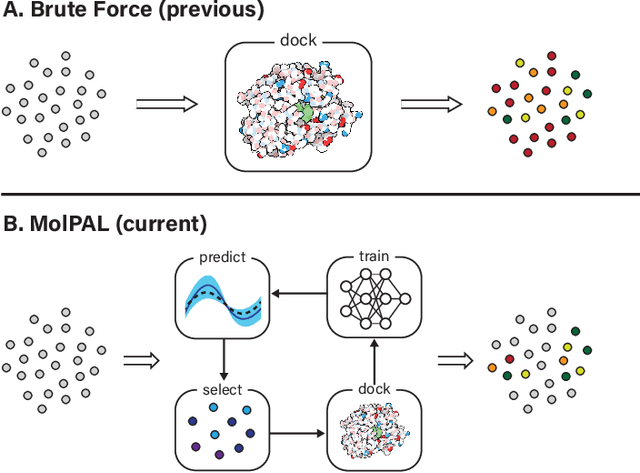


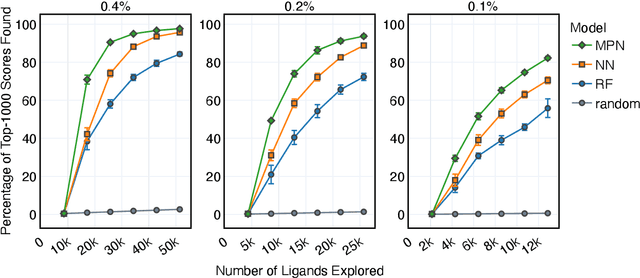
Structure-based virtual screening is an important tool in early stage drug discovery that scores the interactions between a target protein and candidate ligands. As virtual libraries continue to grow (in excess of $10^8$ molecules), so too do the resources necessary to conduct exhaustive virtual screening campaigns on these libraries. However, Bayesian optimization techniques can aid in their exploration: a surrogate structure-property relationship model trained on the predicted affinities of a subset of the library can be applied to the remaining library members, allowing the least promising compounds to be excluded from evaluation. In this study, we assess various surrogate model architectures, acquisition functions, and acquisition batch sizes as applied to several protein-ligand docking datasets and observe significant reductions in computational costs, even when using a greedy acquisition strategy; for example, 87.9% of the top-50000 ligands can be found after testing only 2.4% of a 100M member library. Such model-guided searches mitigate the increasing computational costs of screening increasingly large virtual libraries and can accelerate high-throughput virtual screening campaigns with applications beyond docking.