 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA graph representation of molecular ensembles for polymer property prediction
May 17, 2022
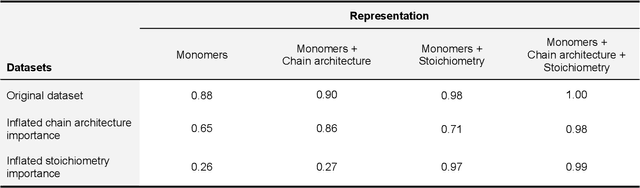
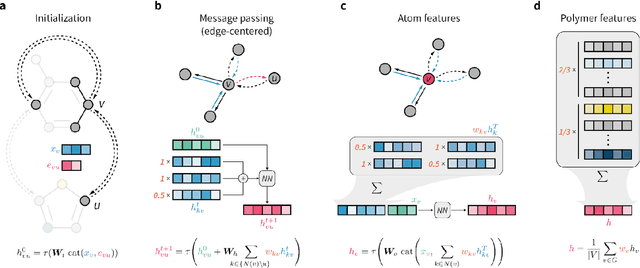

Synthetic polymers are versatile and widely used materials. Similar to small organic molecules, a large chemical space of such materials is hypothetically accessible. Computational property prediction and virtual screening can accelerate polymer design by prioritizing candidates expected to have favorable properties. However, in contrast to organic molecules, polymers are often not well-defined single structures but an ensemble of similar molecules, which poses unique challenges to traditional chemical representations and machine learning approaches. Here, we introduce a graph representation of molecular ensembles and an associated graph neural network architecture that is tailored to polymer property prediction. We demonstrate that this approach captures critical features of polymeric materials, like chain architecture, monomer stoichiometry, and degree of polymerization, and achieves superior accuracy to off-the-shelf cheminformatics methodologies. While doing so, we built a dataset of simulated electron affinity and ionization potential values for >40k polymers with varying monomer composition, stoichiometry, and chain architecture, which may be used in the development of other tailored machine learning approaches. The dataset and machine learning models presented in this work pave the path toward new classes of algorithms for polymer informatics and, more broadly, introduce a framework for the modeling of molecular ensembles.
Self-focusing virtual screening with active design space pruning
May 03, 2022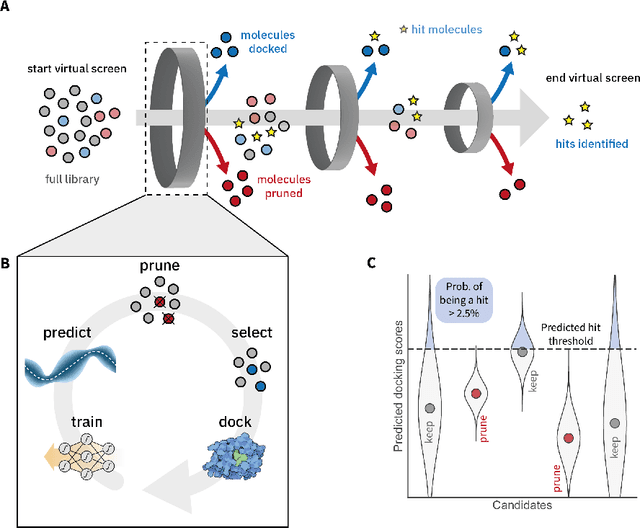
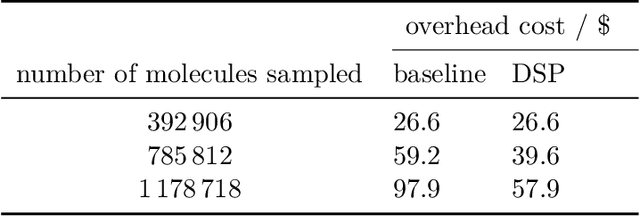
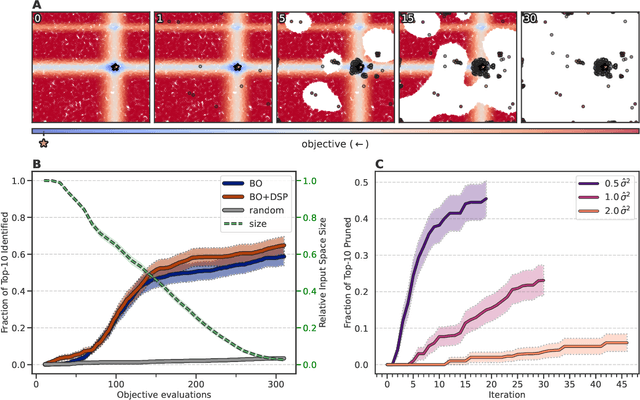
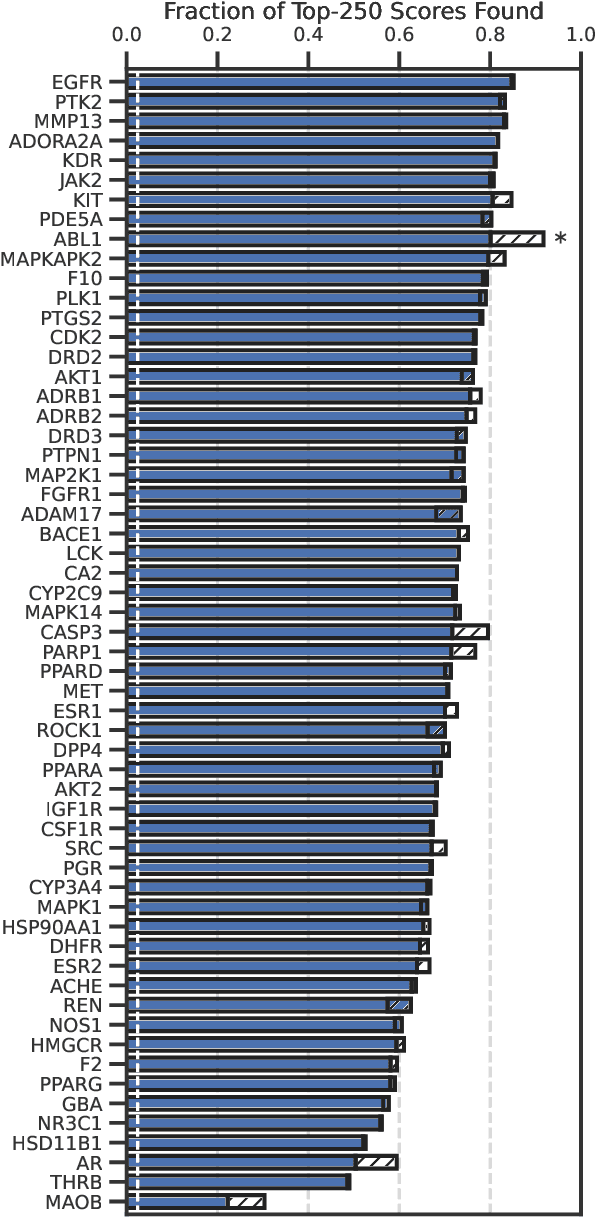
High-throughput virtual screening is an indispensable technique utilized in the discovery of small molecules. In cases where the library of molecules is exceedingly large, the cost of an exhaustive virtual screen may be prohibitive. Model-guided optimization has been employed to lower these costs through dramatic increases in sample efficiency compared to random selection. However, these techniques introduce new costs to the workflow through the surrogate model training and inference steps. In this study, we propose an extension to the framework of model-guided optimization that mitigates inferences costs using a technique we refer to as design space pruning (DSP), which irreversibly removes poor-performing candidates from consideration. We study the application of DSP to a variety of optimization tasks and observe significant reductions in overhead costs while exhibiting similar performance to the baseline optimization. DSP represents an attractive extension of model-guided optimization that can limit overhead costs in optimization settings where these costs are non-negligible relative to objective costs, such as docking.
On scientific understanding with artificial intelligence
Apr 04, 2022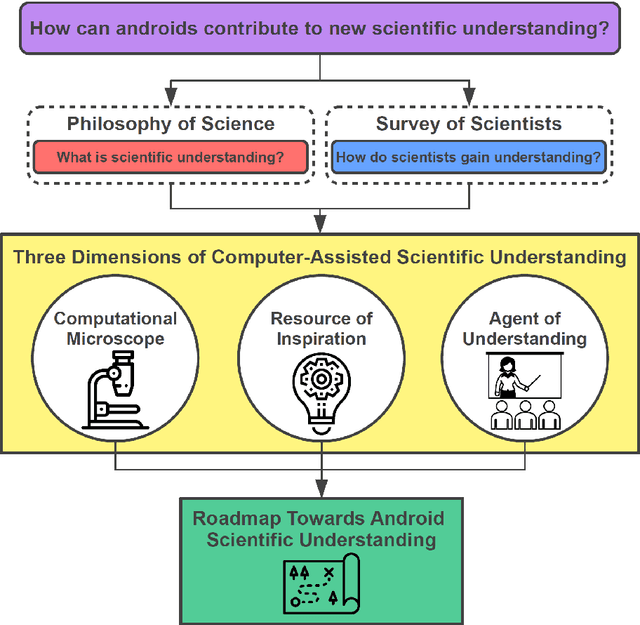
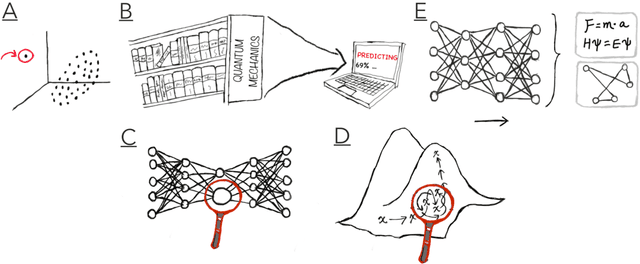
Imagine an oracle that correctly predicts the outcome of every particle physics experiment, the products of every chemical reaction, or the function of every protein. Such an oracle would revolutionize science and technology as we know them. However, as scientists, we would not be satisfied with the oracle itself. We want more. We want to comprehend how the oracle conceived these predictions. This feat, denoted as scientific understanding, has frequently been recognized as the essential aim of science. Now, the ever-growing power of computers and artificial intelligence poses one ultimate question: How can advanced artificial systems contribute to scientific understanding or achieve it autonomously? We are convinced that this is not a mere technical question but lies at the core of science. Therefore, here we set out to answer where we are and where we can go from here. We first seek advice from the philosophy of science to understand scientific understanding. Then we review the current state of the art, both from literature and by collecting dozens of anecdotes from scientists about how they acquired new conceptual understanding with the help of computers. Those combined insights help us to define three dimensions of android-assisted scientific understanding: The android as a I) computational microscope, II) resource of inspiration and the ultimate, not yet existent III) agent of understanding. For each dimension, we explain new avenues to push beyond the status quo and unleash the full power of artificial intelligence's contribution to the central aim of science. We hope our perspective inspires and focuses research towards androids that get new scientific understanding and ultimately bring us closer to true artificial scientists.
Bayesian optimization with known experimental and design constraints for chemistry applications
Mar 29, 2022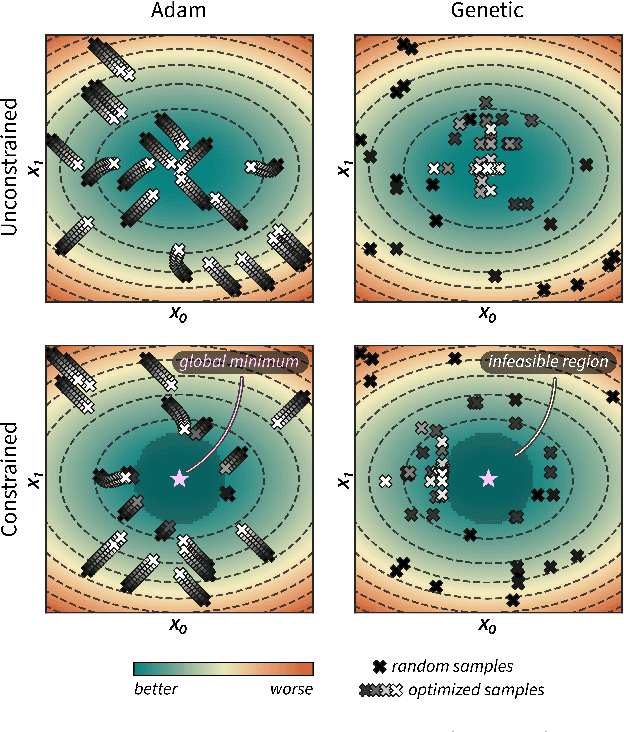
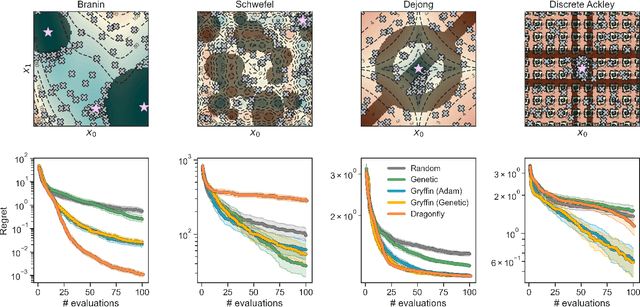

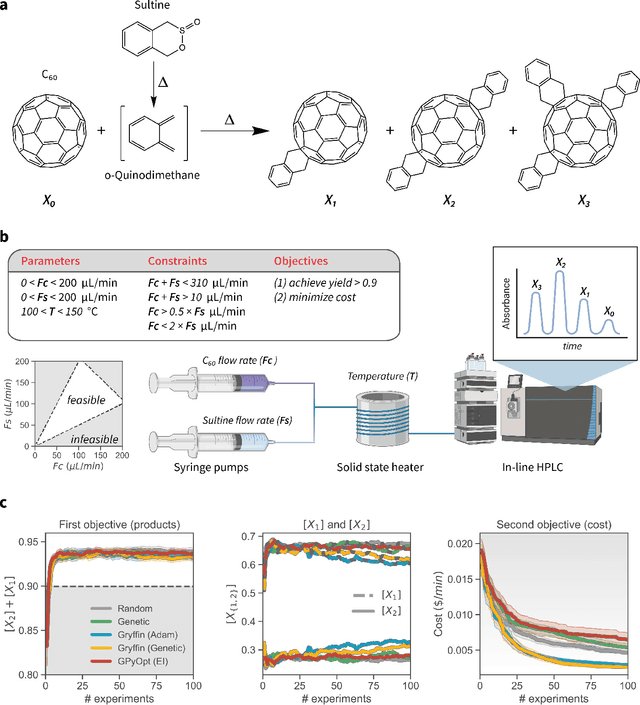
Optimization strategies driven by machine learning, such as Bayesian optimization, are being explored across experimental sciences as an efficient alternative to traditional design of experiment. When combined with automated laboratory hardware and high-performance computing, these strategies enable next-generation platforms for autonomous experimentation. However, the practical application of these approaches is hampered by a lack of flexible software and algorithms tailored to the unique requirements of chemical research. One such aspect is the pervasive presence of constraints in the experimental conditions when optimizing chemical processes or protocols, and in the chemical space that is accessible when designing functional molecules or materials. Although many of these constraints are known a priori, they can be interdependent, non-linear, and result in non-compact optimization domains. In this work, we extend our experiment planning algorithms Phoenics and Gryffin such that they can handle arbitrary known constraints via an intuitive and flexible interface. We benchmark these extended algorithms on continuous and discrete test functions with a diverse set of constraints, demonstrating their flexibility and robustness. In addition, we illustrate their practical utility in two simulated chemical research scenarios: the optimization of the synthesis of o-xylenyl Buckminsterfullerene adducts under constrained flow conditions, and the design of redox active molecules for flow batteries under synthetic accessibility constraints. The tools developed constitute a simple, yet versatile strategy to enable model-based optimization with known experimental constraints, contributing to its applicability as a core component of autonomous platforms for scientific discovery.
Golem: An algorithm for robust experiment and process optimization
Mar 05, 2021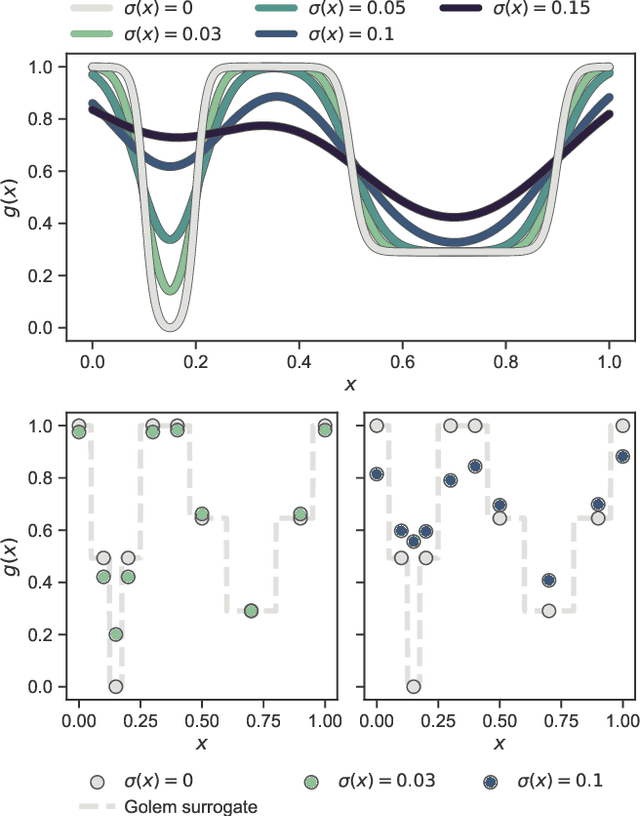
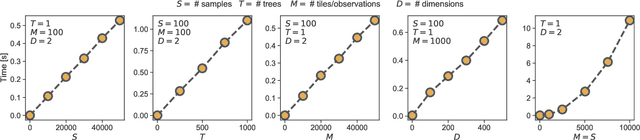
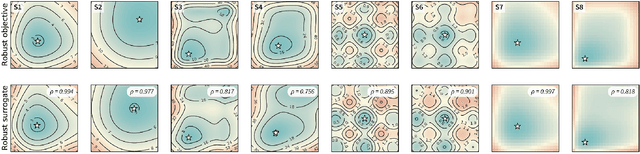
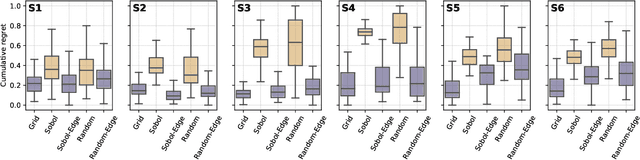
Numerous challenges in science and engineering can be framed as optimization tasks, including the maximization of reaction yields, the optimization of molecular and materials properties, and the fine-tuning of automated hardware protocols. Design of experiment and optimization algorithms are often adopted to solve these tasks efficiently. Increasingly, these experiment planning strategies are coupled with automated hardware to enable autonomous experimental platforms. The vast majority of the strategies used, however, do not consider robustness against the variability of experiment and process conditions. In fact, it is generally assumed that these parameters are exact and reproducible. Yet some experiments may have considerable noise associated with some of their conditions, and process parameters optimized under precise control may be applied in the future under variable operating conditions. In either scenario, the optimal solutions found might not be robust against input variability, affecting the reproducibility of results and returning suboptimal performance in practice. Here, we introduce Golem, an algorithm that is agnostic to the choice of experiment planning strategy and that enables robust experiment and process optimization. Golem identifies optimal solutions that are robust to input uncertainty, thus ensuring the reproducible performance of optimized experimental protocols and processes. It can be used to analyze the robustness of past experiments, or to guide experiment planning algorithms toward robust solutions on the fly. We assess the performance and domain of applicability of Golem through extensive benchmark studies and demonstrate its practical relevance by optimizing an analytical chemistry protocol under the presence of significant noise in its experimental conditions.
Assigning Confidence to Molecular Property Prediction
Feb 23, 2021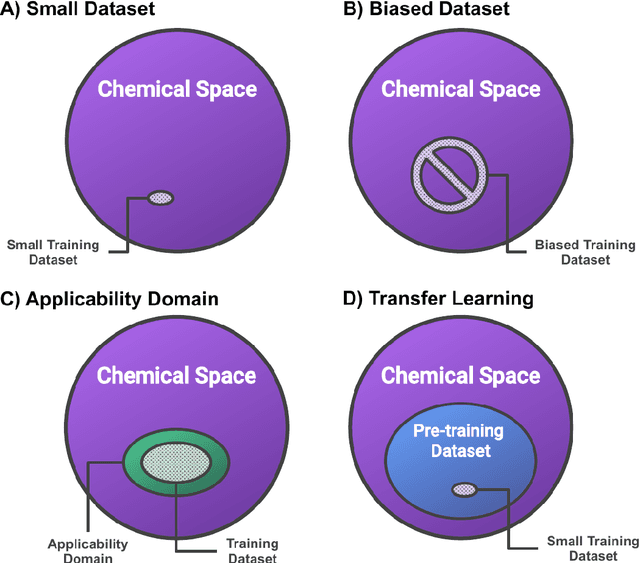
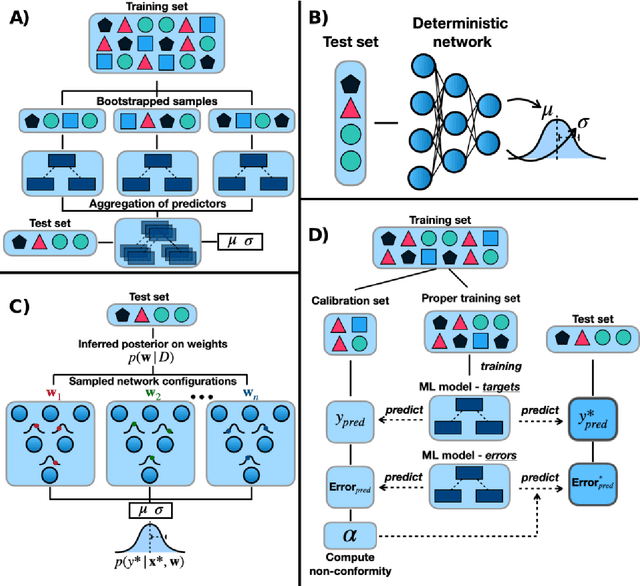
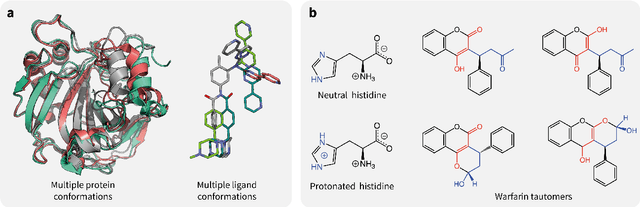
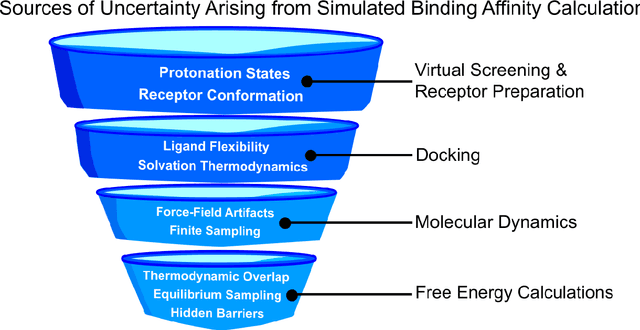
Introduction: Computational modeling has rapidly advanced over the last decades, especially to predict molecular properties for chemistry, material science and drug design. Recently, machine learning techniques have emerged as a powerful and cost-effective strategy to learn from existing datasets and perform predictions on unseen molecules. Accordingly, the explosive rise of data-driven techniques raises an important question: What confidence can be assigned to molecular property predictions and what techniques can be used for that purpose? Areas covered: In this work, we discuss popular strategies for predicting molecular properties relevant to drug design, their corresponding uncertainty sources and methods to quantify uncertainty and confidence. First, our considerations for assessing confidence begin with dataset bias and size, data-driven property prediction and feature design. Next, we discuss property simulation via molecular docking, and free-energy simulations of binding affinity in detail. Lastly, we investigate how these uncertainties propagate to generative models, as they are usually coupled with property predictors. Expert opinion: Computational techniques are paramount to reduce the prohibitive cost and timing of brute-force experimentation when exploring the enormous chemical space. We believe that assessing uncertainty in property prediction models is essential whenever closed-loop drug design campaigns relying on high-throughput virtual screening are deployed. Accordingly, considering sources of uncertainty leads to better-informed experimental validations, more reliable predictions and to more realistic expectations of the entire workflow. Overall, this increases confidence in the predictions and designs and, ultimately, accelerates drug design.
Olympus: a benchmarking framework for noisy optimization and experiment planning
Oct 08, 2020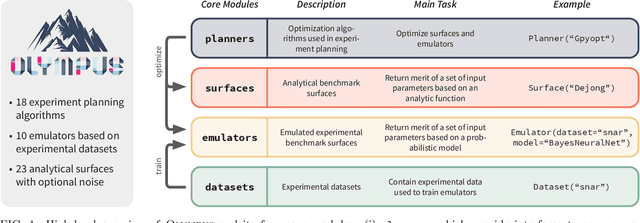
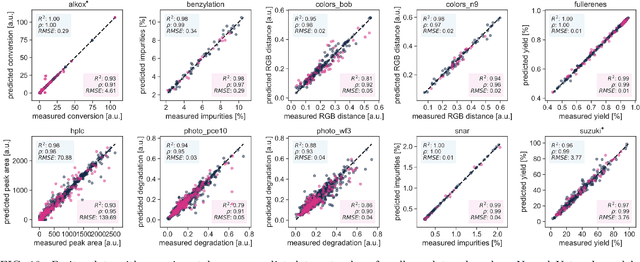

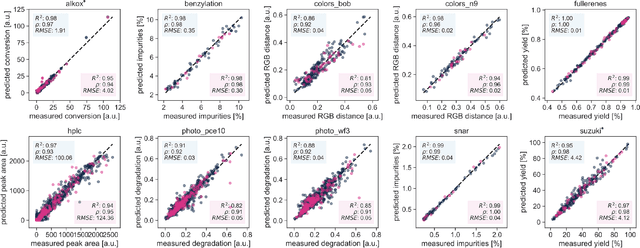
Research challenges encountered across science, engineering, and economics can frequently be formulated as optimization tasks. In chemistry and materials science, recent growth in laboratory digitization and automation has sparked interest in optimization-guided autonomous discovery and closed-loop experimentation. Experiment planning strategies based on off-the-shelf optimization algorithms can be employed in fully autonomous research platforms to achieve desired experimentation goals with the minimum number of trials. However, the experiment planning strategy that is most suitable to a scientific discovery task is a priori unknown while rigorous comparisons of different strategies are highly time and resource demanding. As optimization algorithms are typically benchmarked on low-dimensional synthetic functions, it is unclear how their performance would translate to noisy, higher-dimensional experimental tasks encountered in chemistry and materials science. We introduce Olympus, a software package that provides a consistent and easy-to-use framework for benchmarking optimization algorithms against realistic experiments emulated via probabilistic deep-learning models. Olympus includes a collection of experimentally derived benchmark sets from chemistry and materials science and a suite of experiment planning strategies that can be easily accessed via a user-friendly python interface. Furthermore, Olympus facilitates the integration, testing, and sharing of custom algorithms and user-defined datasets. In brief, Olympus mitigates the barriers associated with benchmarking optimization algorithms on realistic experimental scenarios, promoting data sharing and the creation of a standard framework for evaluating the performance of experiment planning strategies