 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLearning when to observe: A frugal reinforcement learning framework for a high-cost world
Jul 24, 2023



Reinforcement learning (RL) has been shown to learn sophisticated control policies for complex tasks including games, robotics, heating and cooling systems and text generation. The action-perception cycle in RL, however, generally assumes that a measurement of the state of the environment is available at each time step without a cost. In applications such as materials design, deep-sea and planetary robot exploration and medicine, however, there can be a high cost associated with measuring, or even approximating, the state of the environment. In this paper, we survey the recently growing literature that adopts the perspective that an RL agent might not need, or even want, a costly measurement at each time step. Within this context, we propose the Deep Dynamic Multi-Step Observationless Agent (DMSOA), contrast it with the literature and empirically evaluate it on OpenAI gym and Atari Pong environments. Our results, show that DMSOA learns a better policy with fewer decision steps and measurements than the considered alternative from the literature. The corresponding code is available at: \url{https://github.com/cbellinger27/Learning-when-to-observe-in-RL
ChemGymRL: An Interactive Framework for Reinforcement Learning for Digital Chemistry
May 23, 2023



This paper provides a simulated laboratory for making use of Reinforcement Learning (RL) for chemical discovery. Since RL is fairly data intensive, training agents `on-the-fly' by taking actions in the real world is infeasible and possibly dangerous. Moreover, chemical processing and discovery involves challenges which are not commonly found in RL benchmarks and therefore offer a rich space to work in. We introduce a set of highly customizable and open-source RL environments, ChemGymRL, based on the standard Open AI Gym template. ChemGymRL supports a series of interconnected virtual chemical benches where RL agents can operate and train. The paper introduces and details each of these benches using well-known chemical reactions as illustrative examples, and trains a set of standard RL algorithms in each of these benches. Finally, discussion and comparison of the performances of several standard RL methods are provided in addition to a list of directions for future work as a vision for the further development and usage of ChemGymRL.
fintech-kMC: Agent based simulations of financial platforms for design and testing of machine learning systems
Jan 04, 2023



We discuss our simulation tool, fintech-kMC, which is designed to generate synthetic data for machine learning model development and testing. fintech-kMC is an agent-based model driven by a kinetic Monte Carlo (a.k.a. continuous time Monte Carlo) engine which simulates the behaviour of customers using an online digital financial platform. The tool provides an interpretable, reproducible, and realistic way of generating synthetic data which can be used to validate and test AI/ML models and pipelines to be used in real-world customer-facing financial applications.
Training neural networks using Metropolis Monte Carlo and an adaptive variant
May 16, 2022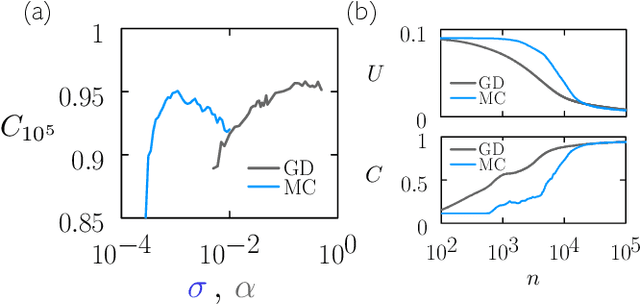
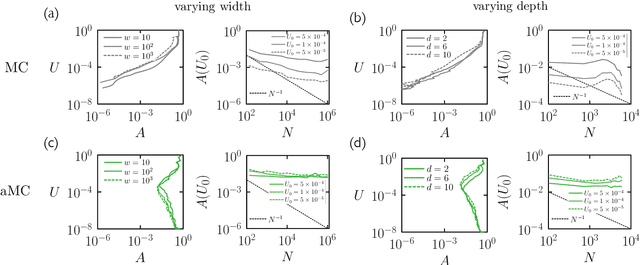
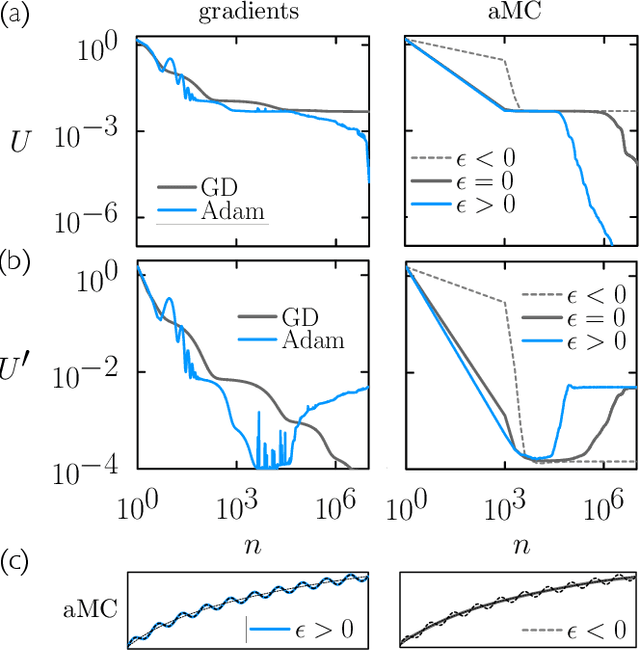
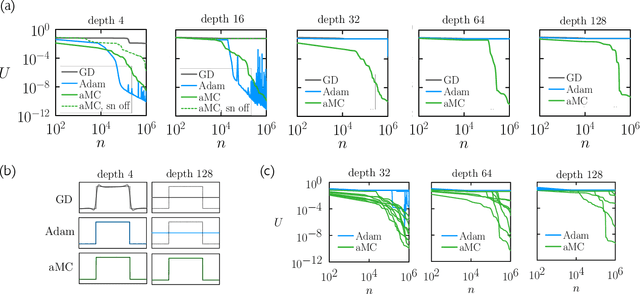
We examine the zero-temperature Metropolis Monte Carlo algorithm as a tool for training a neural network by minimizing a loss function. We find that, as expected on theoretical grounds and shown empirically by other authors, Metropolis Monte Carlo can train a neural net with an accuracy comparable to that of gradient descent, if not necessarily as quickly. The Metropolis algorithm does not fail automatically when the number of parameters of a neural network is large. It can fail when a neural network's structure or neuron activations are strongly heterogenous, and we introduce an adaptive Monte Carlo algorithm, aMC, to overcome these limitations. The intrinsic stochasticity of the Monte Carlo method allows aMC to train neural networks in which the gradient is too small to allow training by gradient descent. We suggest that, as for molecular simulation, Monte Carlo methods offer a complement to gradient-based methods for training neural networks, allowing access to a distinct set of network architectures and principles.
Machine Learning Diffusion Monte Carlo Energy Densities
May 09, 2022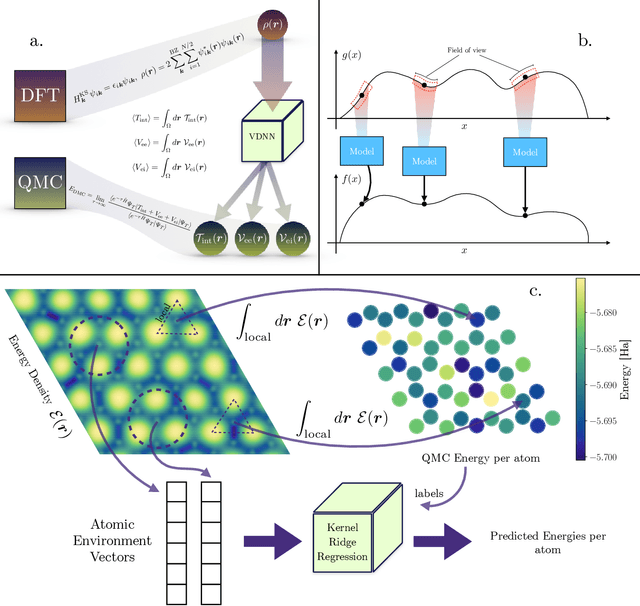
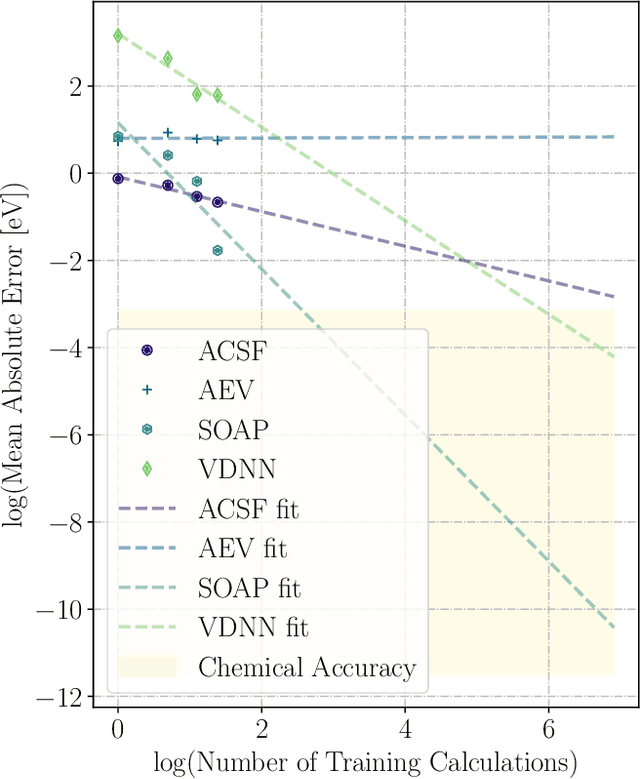
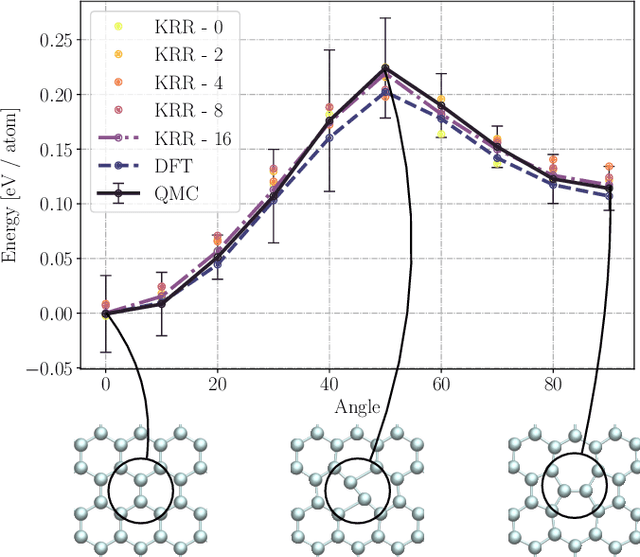
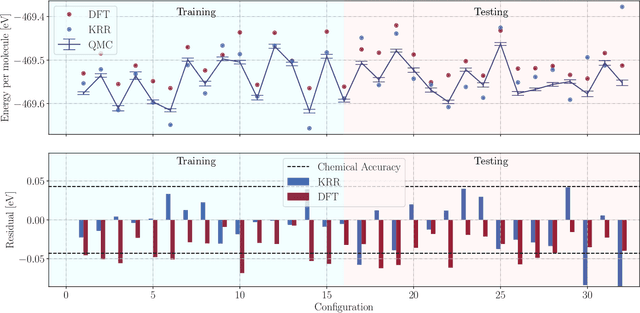
We present two machine learning methodologies which are capable of predicting diffusion Monte Carlo (DMC) energies with small datasets ($\approx$60 DMC calculations in total). The first uses voxel deep neural networks (VDNNs) to predict DMC energy densities using Kohn-Sham density functional theory (DFT) electron densities as input. The second uses kernel ridge regression (KRR) to predict atomic contributions to the DMC total energy using atomic environment vectors as input (we used atom centred symmetry functions, atomic environment vectors from the ANI models, and smooth overlap of atomic positions). We first compare the methodologies on pristine graphene lattices, where we find the KRR methodology performs best in comparison to gradient boosted decision trees, random forest, gaussian process regression, and multilayer perceptrons. In addition, KRR outperforms VDNNs by an order of magnitude. Afterwards, we study the generalizability of KRR to predict the energy barrier associated with a Stone-Wales defect. Lastly, we move from 2D to 3D materials and use KRR to predict total energies of liquid water. In all cases, we find that the KRR models are more accurate than Kohn-Sham DFT and all mean absolute errors are less than chemical accuracy.
Cellular automata can classify data by inducing trajectory phase coexistence
Apr 06, 2022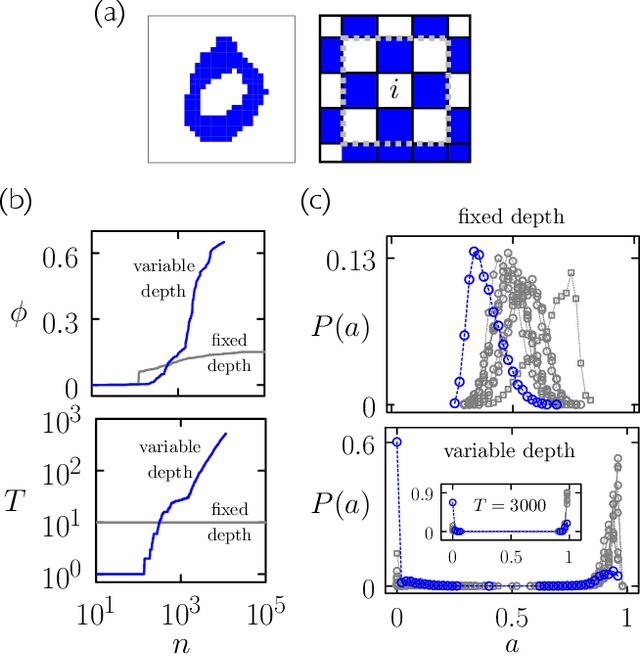
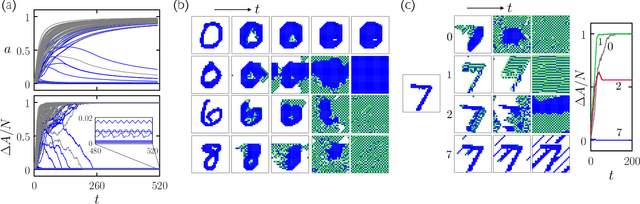
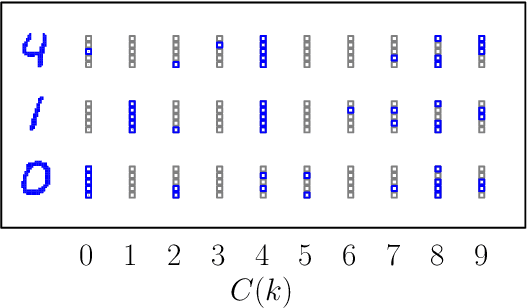
We show that cellular automata can classify data by inducing a form of dynamical phase coexistence. We use Monte Carlo methods to search for general two-dimensional deterministic automata that classify images on the basis of activity, the number of state changes that occur in a trajectory initiated from the image. When the depth of the automaton is a trainable parameter, the search scheme identifies automata that generate a population of dynamical trajectories displaying high or low activity, depending on initial conditions. Automata of this nature behave as nonlinear activation functions with an output that is effectively binary, resembling an emergent version of a spiking neuron. Our work connects machine learning and reservoir computing to phenomena conceptually similar to those seen in physical systems such as magnets and glasses.
Generative Enriched Sequential Learning (ESL) Approach for Molecular Design via Augmented Domain Knowledge
Apr 05, 2022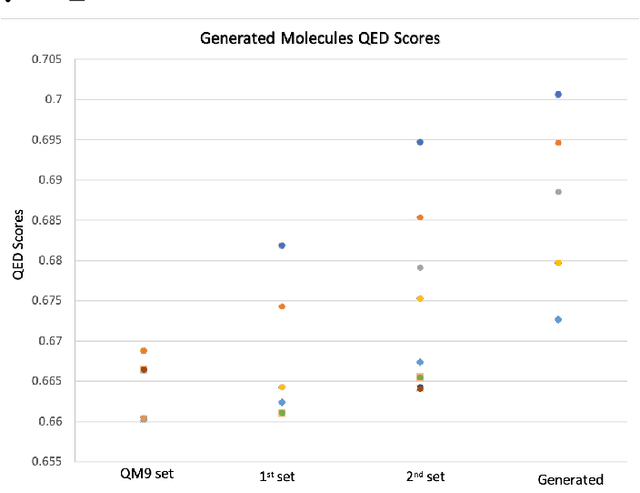
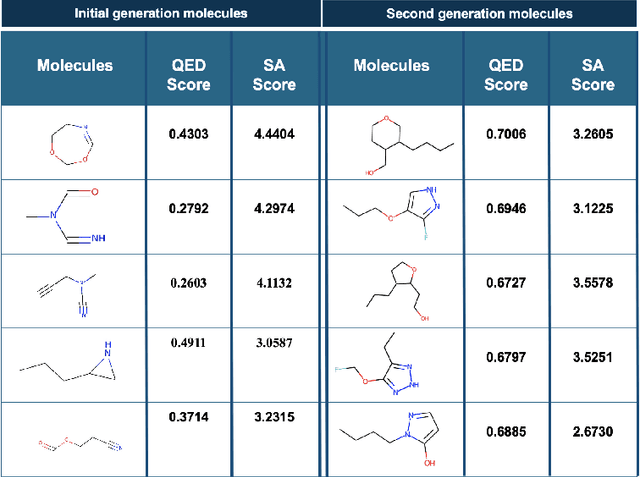
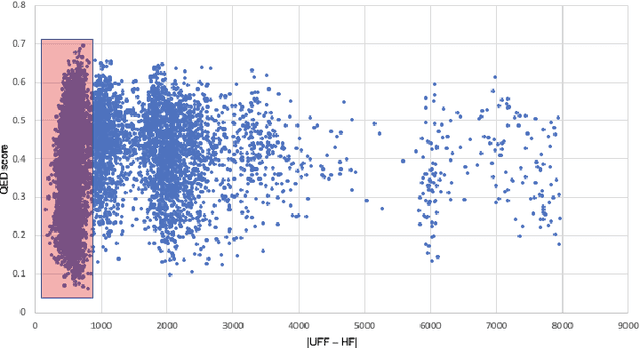
Deploying generative machine learning techniques to generate novel chemical structures based on molecular fingerprint representation has been well established in molecular design. Typically, sequential learning (SL) schemes such as hidden Markov models (HMM) and, more recently, in the sequential deep learning context, recurrent neural network (RNN) and long short-term memory (LSTM) were used extensively as generative models to discover unprecedented molecules. To this end, emission probability between two states of atoms plays a central role without considering specific chemical or physical properties. Lack of supervised domain knowledge can mislead the learning procedure to be relatively biased to the prevalent molecules observed in the training data that are not necessarily of interest. We alleviated this drawback by augmenting the training data with domain knowledge, e.g. quantitative estimates of the drug-likeness score (QEDs). As such, our experiments demonstrated that with this subtle trick called enriched sequential learning (ESL), specific patterns of particular interest can be learnt better, which led to generating de novo molecules with ameliorated QEDs.
Learning stochastic dynamics and predicting emergent behavior using transformers
Feb 17, 2022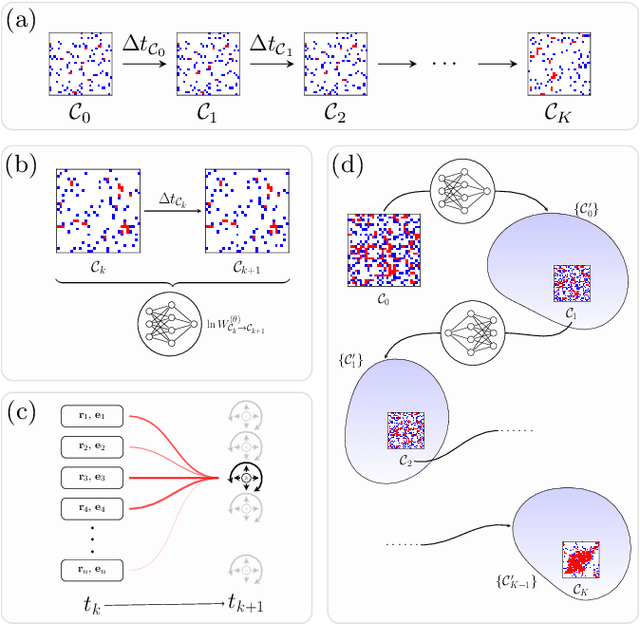
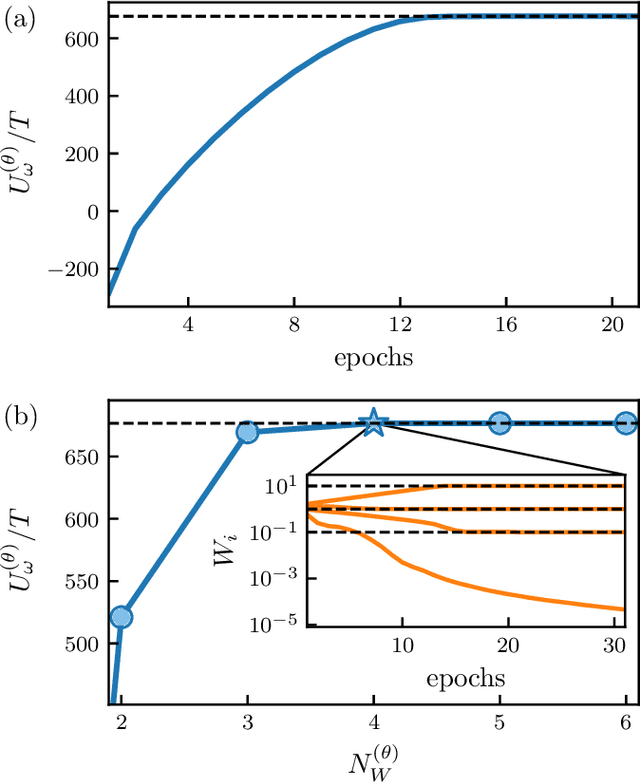
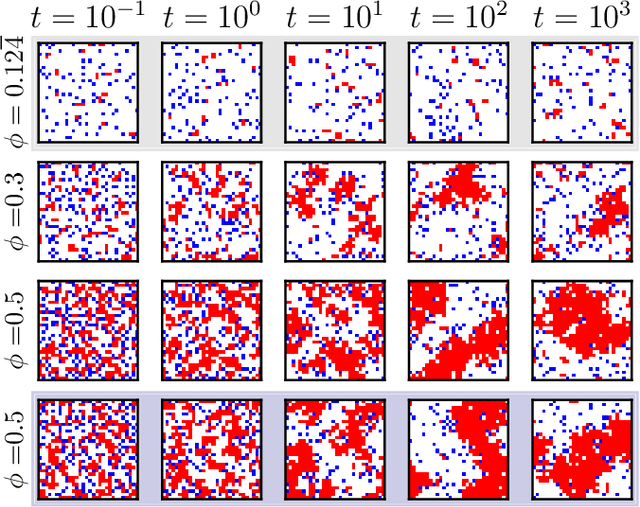
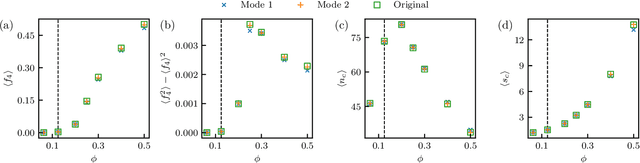
We show that a neural network originally designed for language processing can learn the dynamical rules of a stochastic system by observation of a single dynamical trajectory of the system, and can accurately predict its emergent behavior under conditions not observed during training. We consider a lattice model of active matter undergoing continuous-time Monte Carlo dynamics, simulated at a density at which its steady state comprises small, dispersed clusters. We train a neural network called a transformer on a single trajectory of the model. The transformer, which we show has the capacity to represent dynamical rules that are numerous and nonlocal, learns that the dynamics of this model consists of a small number of processes. Forward-propagated trajectories of the trained transformer, at densities not encountered during training, exhibit motility-induced phase separation and so predict the existence of a nonequilibrium phase transition. Transformers have the flexibility to learn dynamical rules from observation without explicit enumeration of rates or coarse-graining of configuration space, and so the procedure used here can be applied to a wide range of physical systems, including those with large and complex dynamical generators.
Dynamic programming with partial information to overcome navigational uncertainty in a nautical environment
Dec 29, 2021
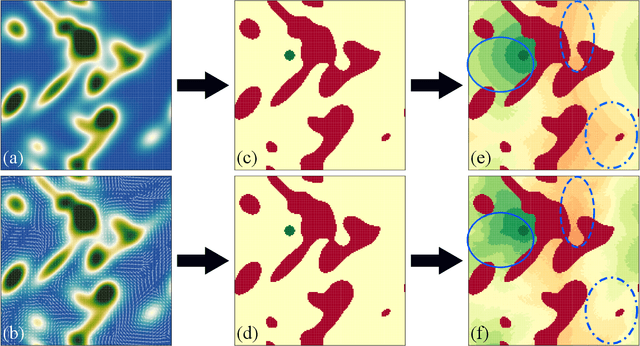
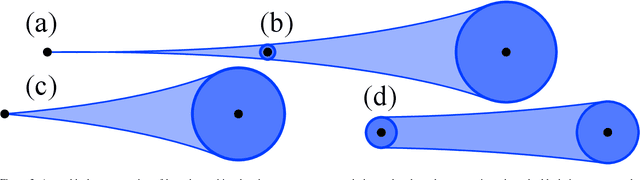
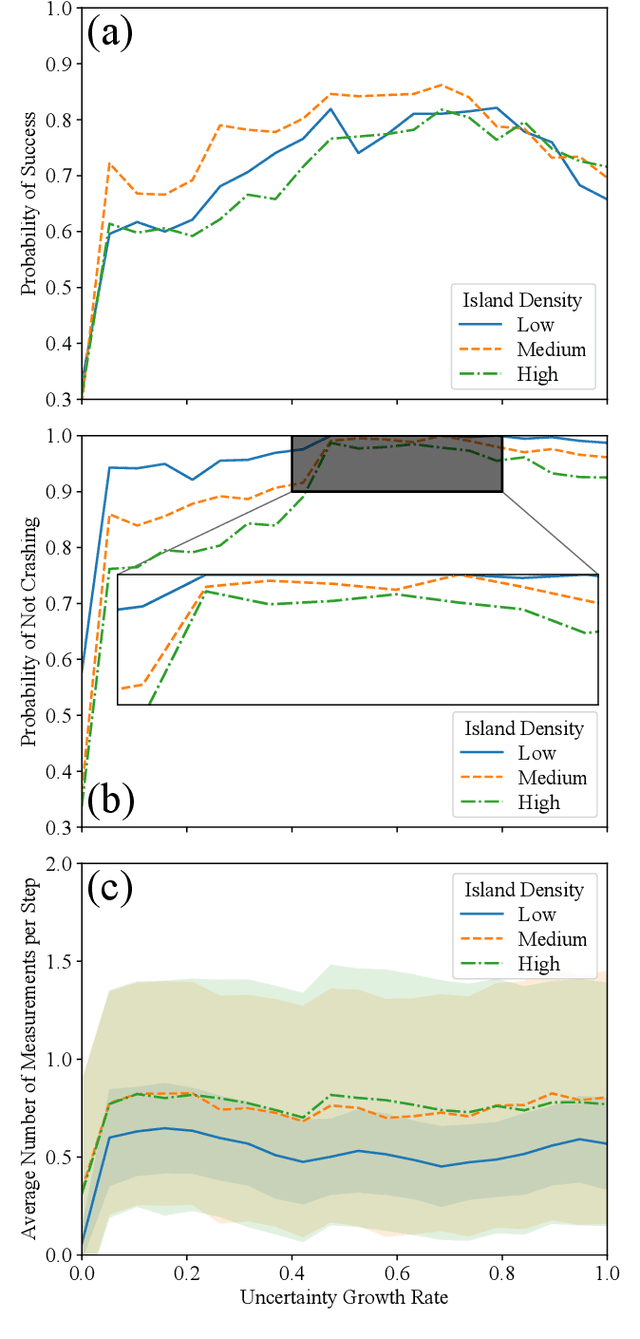
Using a toy nautical navigation environment, we show that dynamic programming can be used when only partial information about a partially observed Markov decision process (POMDP) is known. By incorporating uncertainty into our model, we show that navigation policies can be constructed that maintain safety. Adding controlled sensing methods, we show that these policies can also lower measurement costs at the same time.
Scientific Discovery and the Cost of Measurement -- Balancing Information and Cost in Reinforcement Learning
Dec 14, 2021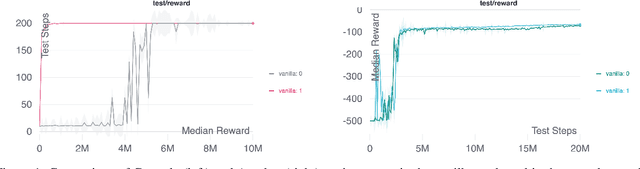
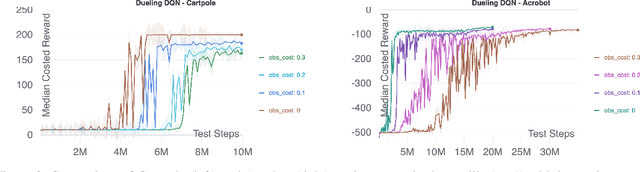
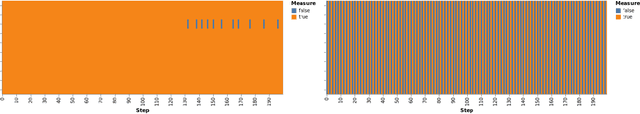
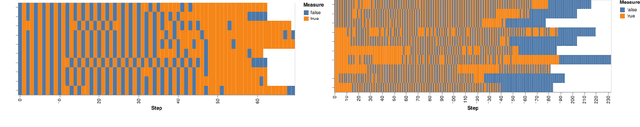
The use of reinforcement learning (RL) in scientific applications, such as materials design and automated chemistry, is increasing. A major challenge, however, lies in fact that measuring the state of the system is often costly and time consuming in scientific applications, whereas policy learning with RL requires a measurement after each time step. In this work, we make the measurement costs explicit in the form of a costed reward and propose a framework that enables off-the-shelf deep RL algorithms to learn a policy for both selecting actions and determining whether or not to measure the current state of the system at each time step. In this way, the agents learn to balance the need for information with the cost of information. Our results show that when trained under this regime, the Dueling DQN and PPO agents can learn optimal action policies whilst making up to 50\% fewer state measurements, and recurrent neural networks can produce a greater than 50\% reduction in measurements. We postulate the these reduction can help to lower the barrier to applying RL to real-world scientific applications.