 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGuided Multi-objective Generative AI to Enhance Structure-based Drug Design
May 20, 2024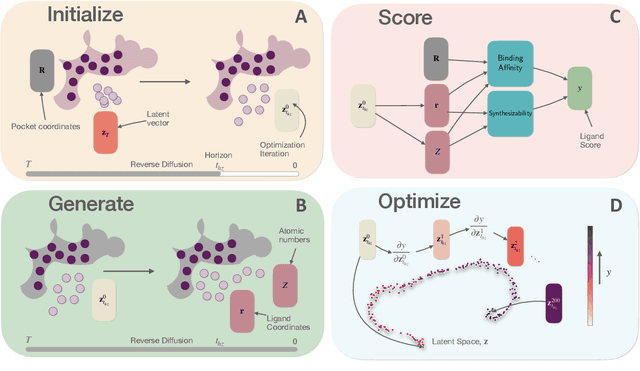

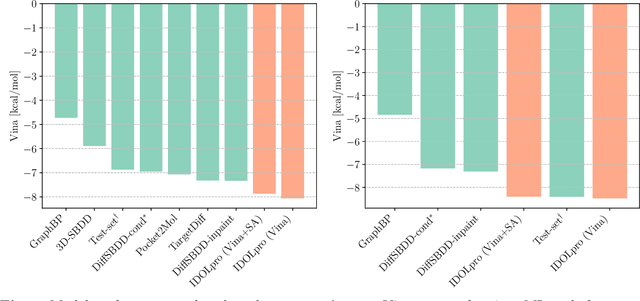
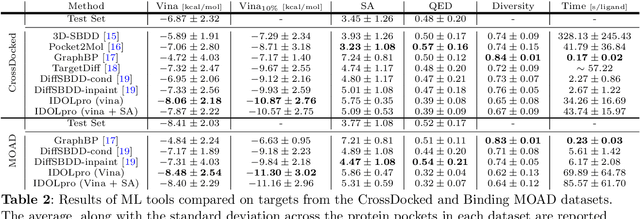
Generative AI has the potential to revolutionize drug discovery. Yet, despite recent advances in machine learning, existing models cannot generate molecules that satisfy all desired physicochemical properties. Herein, we describe IDOLpro, a novel generative chemistry AI combining deep diffusion with multi-objective optimization for structure-based drug design. The latent variables of the diffusion model are guided by differentiable scoring functions to explore uncharted chemical space and generate novel ligands in silico, optimizing a plurality of target physicochemical properties. We demonstrate its effectiveness by generating ligands with optimized binding affinity and synthetic accessibility on two benchmark sets. IDOLpro produces ligands with binding affinities over 10% higher than the next best state-of-the-art on each test set. On a test set of experimental complexes, IDOLpro is the first to surpass the performance of experimentally observed ligands. IDOLpro can accommodate other scoring functions (e.g. ADME-Tox) to accelerate hit-finding, hit-to-lead, and lead optimization for drug discovery.
Machine Learning Diffusion Monte Carlo Energy Densities
May 09, 2022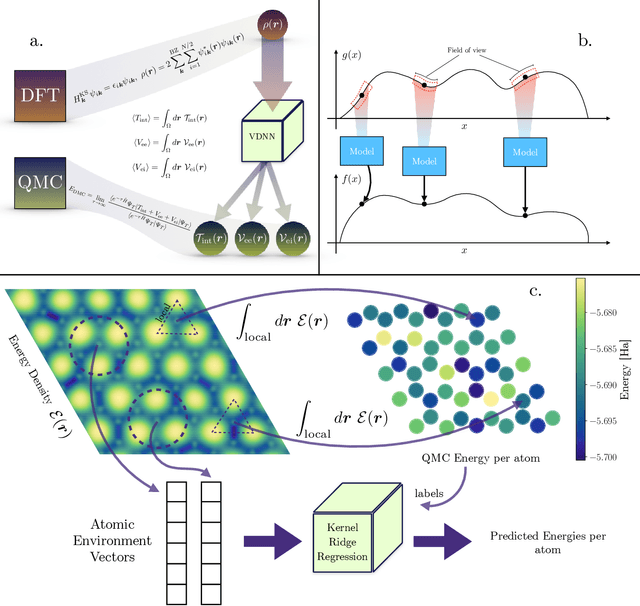
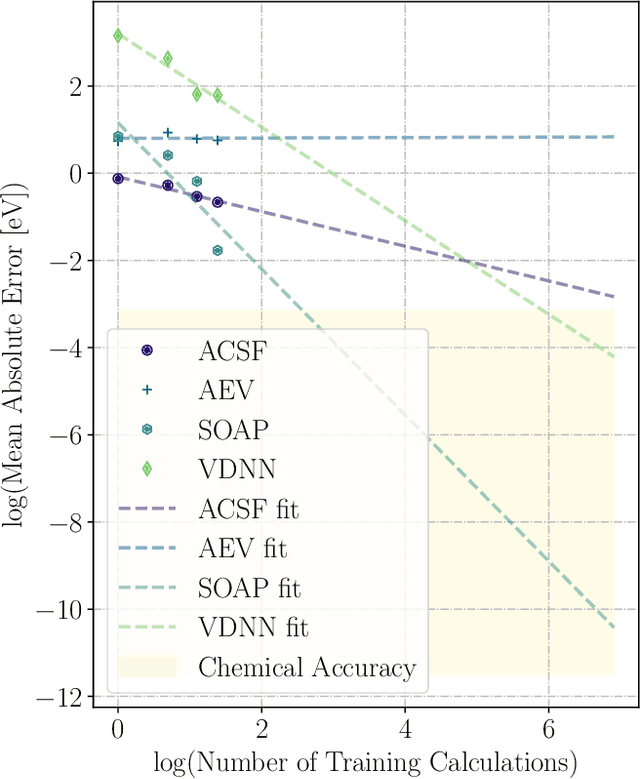
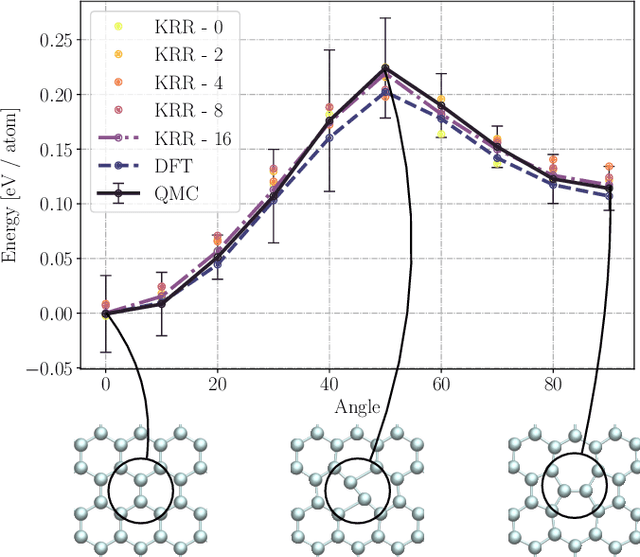
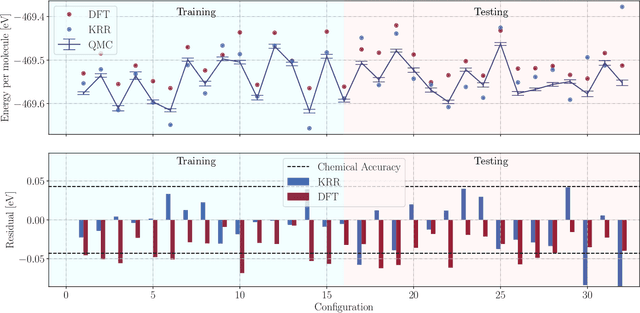
We present two machine learning methodologies which are capable of predicting diffusion Monte Carlo (DMC) energies with small datasets ($\approx$60 DMC calculations in total). The first uses voxel deep neural networks (VDNNs) to predict DMC energy densities using Kohn-Sham density functional theory (DFT) electron densities as input. The second uses kernel ridge regression (KRR) to predict atomic contributions to the DMC total energy using atomic environment vectors as input (we used atom centred symmetry functions, atomic environment vectors from the ANI models, and smooth overlap of atomic positions). We first compare the methodologies on pristine graphene lattices, where we find the KRR methodology performs best in comparison to gradient boosted decision trees, random forest, gaussian process regression, and multilayer perceptrons. In addition, KRR outperforms VDNNs by an order of magnitude. Afterwards, we study the generalizability of KRR to predict the energy barrier associated with a Stone-Wales defect. Lastly, we move from 2D to 3D materials and use KRR to predict total energies of liquid water. In all cases, we find that the KRR models are more accurate than Kohn-Sham DFT and all mean absolute errors are less than chemical accuracy.
Twin Neural Network Regression
Dec 29, 2020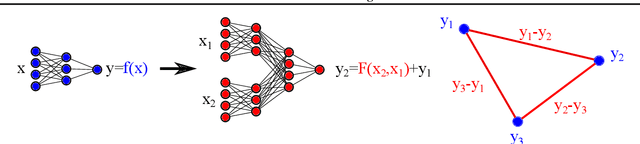
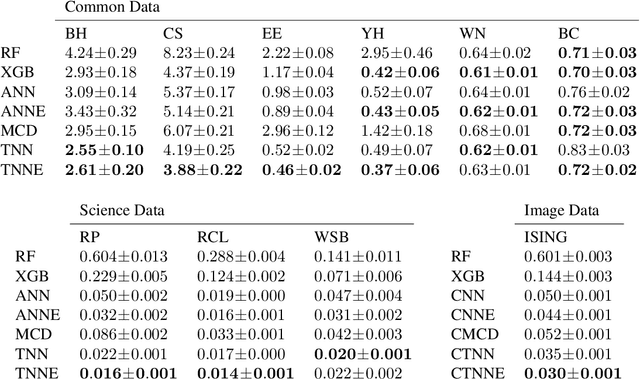
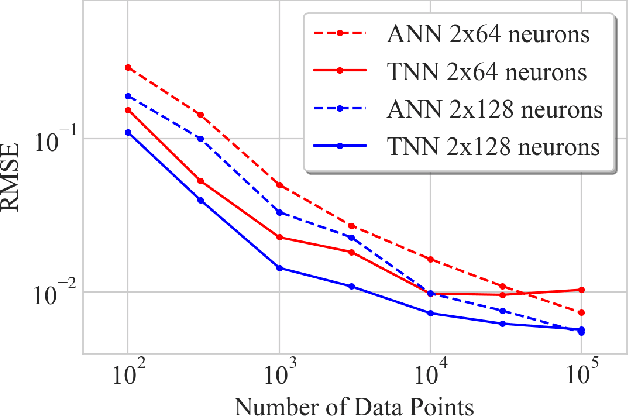
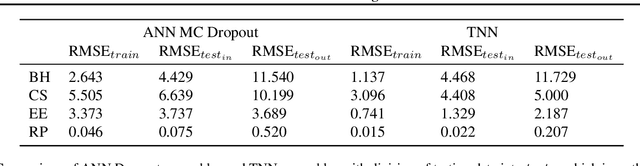
We introduce twin neural network (TNN) regression. This method predicts differences between the target values of two different data points rather than the targets themselves. The solution of a traditional regression problem is then obtained by averaging over an ensemble of all predicted differences between the targets of an unseen data point and all training data points. Whereas ensembles are normally costly to produce, TNN regression intrinsically creates an ensemble of predictions of twice the size of the training set while only training a single neural network. Since ensembles have been shown to be more accurate than single models this property naturally transfers to TNN regression. We show that TNNs are able to compete or yield more accurate predictions for different data sets, compared to other state-of-the-art methods. Furthermore, TNN regression is constrained by self-consistency conditions. We find that the violation of these conditions provides an estimate for the prediction uncertainty.