 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMolGuidance: Advanced Guidance Strategies for Conditional Molecular Generation with Flow Matching
Dec 13, 2025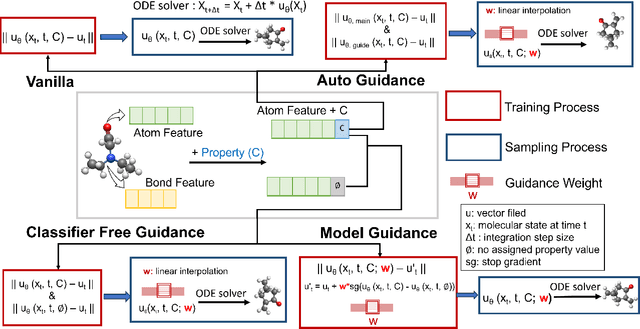

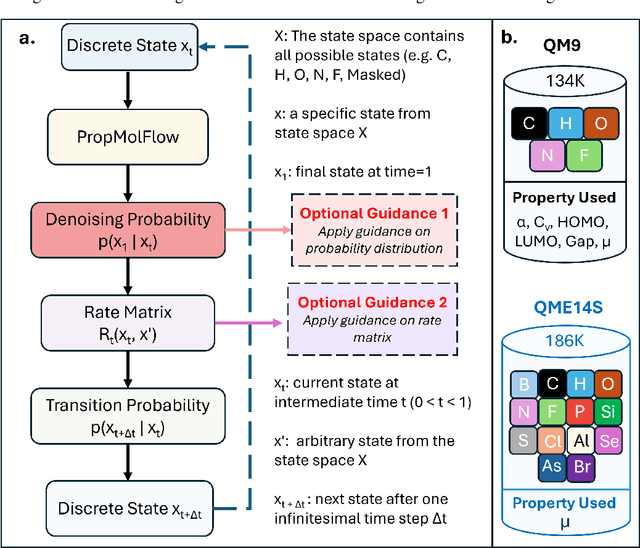
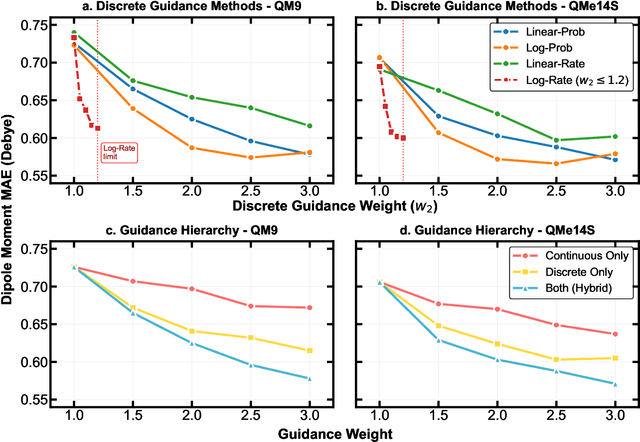
Key objectives in conditional molecular generation include ensuring chemical validity, aligning generated molecules with target properties, promoting structural diversity, and enabling efficient sampling for discovery. Recent advances in computer vision introduced a range of new guidance strategies for generative models, many of which can be adapted to support these goals. In this work, we integrate state-of-the-art guidance methods -- including classifier-free guidance, autoguidance, and model guidance -- in a leading molecule generation framework built on an SE(3)-equivariant flow matching process. We propose a hybrid guidance strategy that separately guides continuous and discrete molecular modalities -- operating on velocity fields and predicted logits, respectively -- while jointly optimizing their guidance scales via Bayesian optimization. Our implementation, benchmarked on the QM9 and QMe14S datasets, achieves new state-of-the-art performance in property alignment for de novo molecular generation. The generated molecules also exhibit high structural validity. Furthermore, we systematically compare the strengths and limitations of various guidance methods, offering insights into their broader applicability.
Open Materials Generation with Stochastic Interpolants
Feb 04, 2025The discovery of new materials is essential for enabling technological advancements. Computational approaches for predicting novel materials must effectively learn the manifold of stable crystal structures within an infinite design space. We introduce Open Materials Generation (OMG), a unifying framework for the generative design and discovery of inorganic crystalline materials. OMG employs stochastic interpolants (SI) to bridge an arbitrary base distribution to the target distribution of inorganic crystals via a broad class of tunable stochastic processes, encompassing both diffusion models and flow matching as special cases. In this work, we adapt the SI framework by integrating an equivariant graph representation of crystal structures and extending it to account for periodic boundary conditions in unit cell representations. Additionally, we couple the SI flow over spatial coordinates and lattice vectors with discrete flow matching for atomic species. We benchmark OMG's performance on two tasks: Crystal Structure Prediction (CSP) for specified compositions, and 'de novo' generation (DNG) aimed at discovering stable, novel, and unique structures. In our ground-up implementation of OMG, we refine and extend both CSP and DNG metrics compared to previous works. OMG establishes a new state-of-the-art in generative modeling for materials discovery, outperforming purely flow-based and diffusion-based implementations. These results underscore the importance of designing flexible deep learning frameworks to accelerate progress in materials science.
Guided Multi-objective Generative AI to Enhance Structure-based Drug Design
May 20, 2024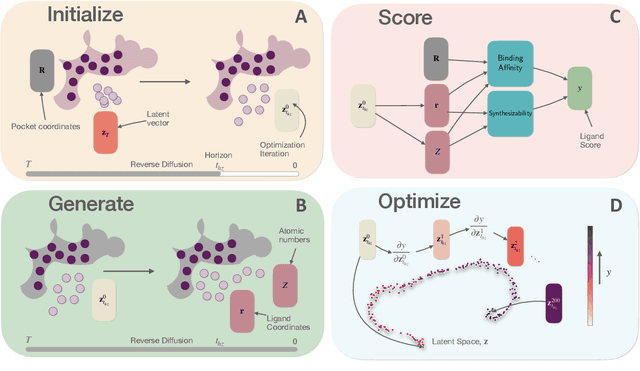

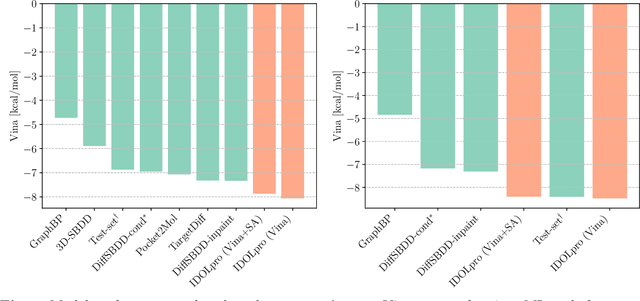
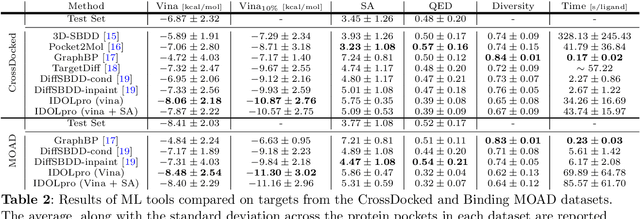
Generative AI has the potential to revolutionize drug discovery. Yet, despite recent advances in machine learning, existing models cannot generate molecules that satisfy all desired physicochemical properties. Herein, we describe IDOLpro, a novel generative chemistry AI combining deep diffusion with multi-objective optimization for structure-based drug design. The latent variables of the diffusion model are guided by differentiable scoring functions to explore uncharted chemical space and generate novel ligands in silico, optimizing a plurality of target physicochemical properties. We demonstrate its effectiveness by generating ligands with optimized binding affinity and synthetic accessibility on two benchmark sets. IDOLpro produces ligands with binding affinities over 10% higher than the next best state-of-the-art on each test set. On a test set of experimental complexes, IDOLpro is the first to surpass the performance of experimentally observed ligands. IDOLpro can accommodate other scoring functions (e.g. ADME-Tox) to accelerate hit-finding, hit-to-lead, and lead optimization for drug discovery.