 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAmplifying Pathological Detection in EEG Signaling Pathways through Cross-Dataset Transfer Learning
Sep 19, 2023


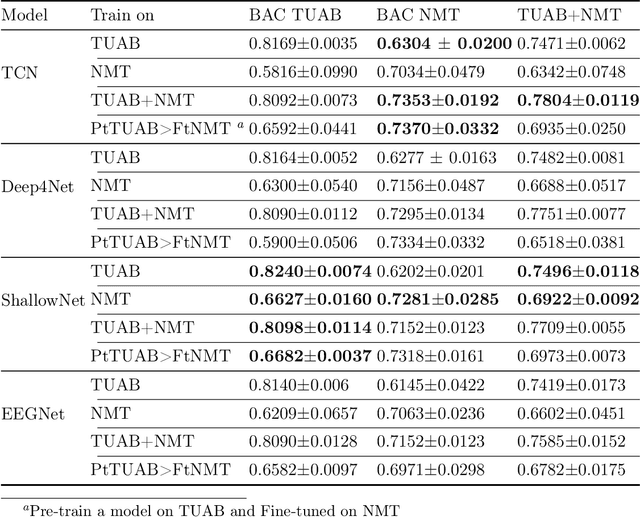
Pathology diagnosis based on EEG signals and decoding brain activity holds immense importance in understanding neurological disorders. With the advancement of artificial intelligence methods and machine learning techniques, the potential for accurate data-driven diagnoses and effective treatments has grown significantly. However, applying machine learning algorithms to real-world datasets presents diverse challenges at multiple levels. The scarcity of labelled data, especially in low regime scenarios with limited availability of real patient cohorts due to high costs of recruitment, underscores the vital deployment of scaling and transfer learning techniques. In this study, we explore a real-world pathology classification task to highlight the effectiveness of data and model scaling and cross-dataset knowledge transfer. As such, we observe varying performance improvements through data scaling, indicating the need for careful evaluation and labelling. Additionally, we identify the challenges of possible negative transfer and emphasize the significance of some key components to overcome distribution shifts and potential spurious correlations and achieve positive transfer. We see improvement in the performance of the target model on the target (NMT) datasets by using the knowledge from the source dataset (TUAB) when a low amount of labelled data was available. Our findings indicate a small and generic model (e.g. ShallowNet) performs well on a single dataset, however, a larger model (e.g. TCN) performs better on transfer and learning from a larger and diverse dataset.
Machine learning for the prediction of safe and biologically active organophosphorus molecules
Feb 21, 2023


Drug discovery is a complex process with a large molecular space to be considered. By constraining the search space, the fragment-based drug design is an approach that can effectively sample the chemical space of interest. Here we propose a framework of Recurrent Neural Networks (RNN) with an attention model to sample the chemical space of organophosphorus molecules using the fragment-based approach. The framework is trained with a ZINC dataset that is screened for high druglikeness scores. The goal is to predict molecules with similar biological action modes as organophosphorus pesticides or chemical warfare agents yet less toxic to humans. The generated molecules contain a starting fragment of PO2F but have a bulky hydrocarbon side chain limiting its binding effectiveness to the targeted protein.
CHA2: CHemistry Aware Convex Hull Autoencoder Towards Inverse Molecular Design
Feb 21, 2023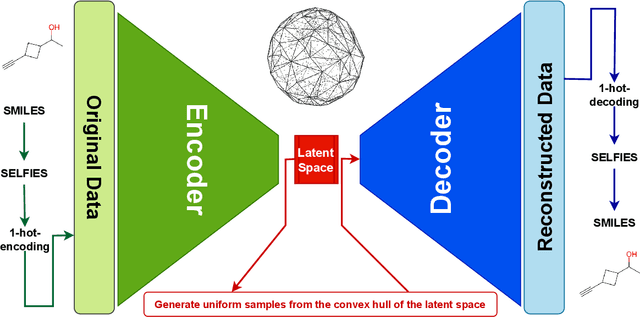
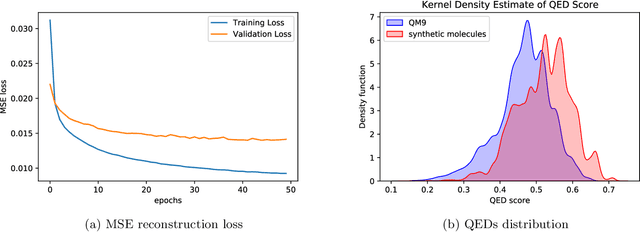


Optimizing molecular design and discovering novel chemical structures to meet certain objectives, such as quantitative estimates of the drug-likeness score (QEDs), is NP-hard due to the vast combinatorial design space of discrete molecular structures, which makes it near impossible to explore the entire search space comprehensively to exploit de novo structures with properties of interest. To address this challenge, reducing the intractable search space into a lower-dimensional latent volume helps examine molecular candidates more feasibly via inverse design. Autoencoders are suitable deep learning techniques, equipped with an encoder that reduces the discrete molecular structure into a latent space and a decoder that inverts the search space back to the molecular design. The continuous property of the latent space, which characterizes the discrete chemical structures, provides a flexible representation for inverse design in order to discover novel molecules. However, exploring this latent space requires certain insights to generate new structures. We propose using a convex hall surrounding the top molecules in terms of high QEDs to ensnare a tight subspace in the latent representation as an efficient way to reveal novel molecules with high QEDs. We demonstrate the effectiveness of our suggested method by using the QM9 as a training dataset along with the Self- Referencing Embedded Strings (SELFIES) representation to calibrate the autoencoder in order to carry out the Inverse molecular design that leads to unfold novel chemical structure.
Generative Enriched Sequential Learning (ESL) Approach for Molecular Design via Augmented Domain Knowledge
Apr 05, 2022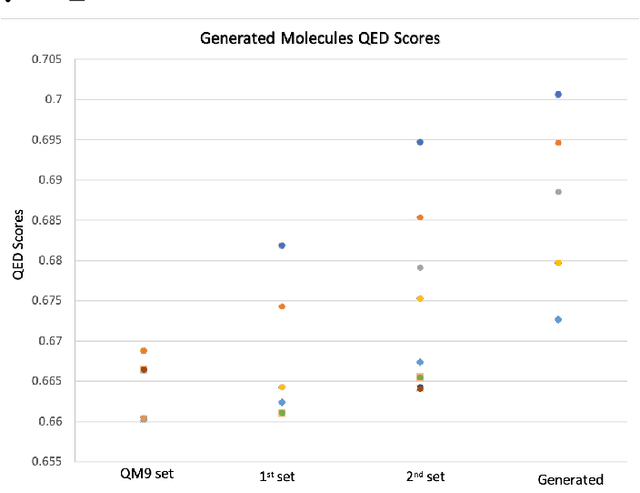
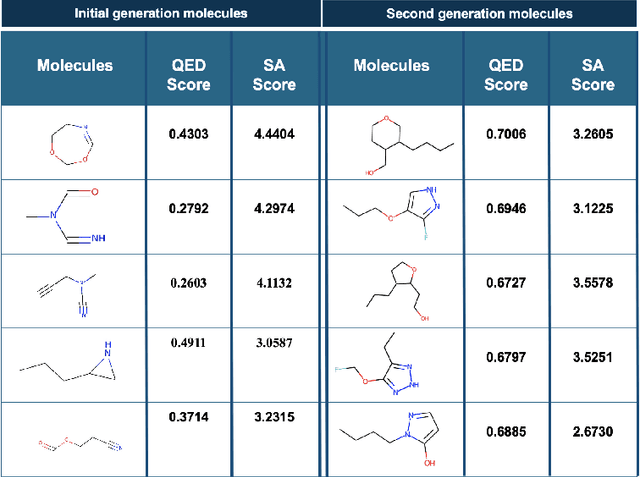
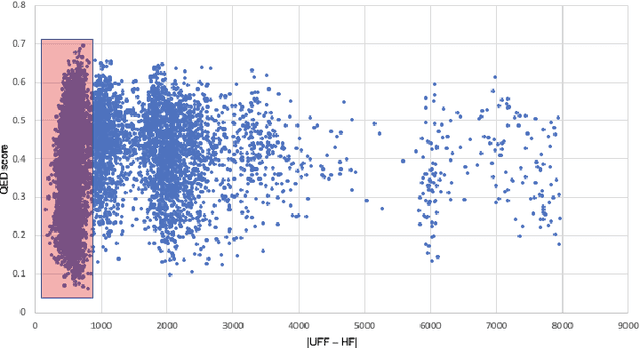
Deploying generative machine learning techniques to generate novel chemical structures based on molecular fingerprint representation has been well established in molecular design. Typically, sequential learning (SL) schemes such as hidden Markov models (HMM) and, more recently, in the sequential deep learning context, recurrent neural network (RNN) and long short-term memory (LSTM) were used extensively as generative models to discover unprecedented molecules. To this end, emission probability between two states of atoms plays a central role without considering specific chemical or physical properties. Lack of supervised domain knowledge can mislead the learning procedure to be relatively biased to the prevalent molecules observed in the training data that are not necessarily of interest. We alleviated this drawback by augmenting the training data with domain knowledge, e.g. quantitative estimates of the drug-likeness score (QEDs). As such, our experiments demonstrated that with this subtle trick called enriched sequential learning (ESL), specific patterns of particular interest can be learnt better, which led to generating de novo molecules with ameliorated QEDs.