 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCHA2: CHemistry Aware Convex Hull Autoencoder Towards Inverse Molecular Design
Feb 21, 2023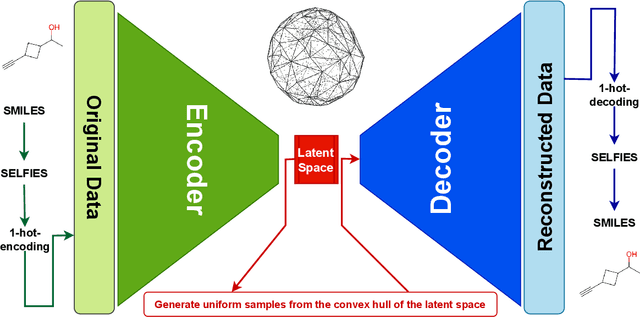
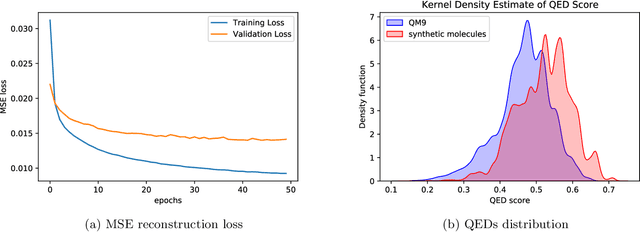


Optimizing molecular design and discovering novel chemical structures to meet certain objectives, such as quantitative estimates of the drug-likeness score (QEDs), is NP-hard due to the vast combinatorial design space of discrete molecular structures, which makes it near impossible to explore the entire search space comprehensively to exploit de novo structures with properties of interest. To address this challenge, reducing the intractable search space into a lower-dimensional latent volume helps examine molecular candidates more feasibly via inverse design. Autoencoders are suitable deep learning techniques, equipped with an encoder that reduces the discrete molecular structure into a latent space and a decoder that inverts the search space back to the molecular design. The continuous property of the latent space, which characterizes the discrete chemical structures, provides a flexible representation for inverse design in order to discover novel molecules. However, exploring this latent space requires certain insights to generate new structures. We propose using a convex hall surrounding the top molecules in terms of high QEDs to ensnare a tight subspace in the latent representation as an efficient way to reveal novel molecules with high QEDs. We demonstrate the effectiveness of our suggested method by using the QM9 as a training dataset along with the Self- Referencing Embedded Strings (SELFIES) representation to calibrate the autoencoder in order to carry out the Inverse molecular design that leads to unfold novel chemical structure.
Machine learning for the prediction of safe and biologically active organophosphorus molecules
Feb 21, 2023


Drug discovery is a complex process with a large molecular space to be considered. By constraining the search space, the fragment-based drug design is an approach that can effectively sample the chemical space of interest. Here we propose a framework of Recurrent Neural Networks (RNN) with an attention model to sample the chemical space of organophosphorus molecules using the fragment-based approach. The framework is trained with a ZINC dataset that is screened for high druglikeness scores. The goal is to predict molecules with similar biological action modes as organophosphorus pesticides or chemical warfare agents yet less toxic to humans. The generated molecules contain a starting fragment of PO2F but have a bulky hydrocarbon side chain limiting its binding effectiveness to the targeted protein.