 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA graph representation of molecular ensembles for polymer property prediction
Paper and Code

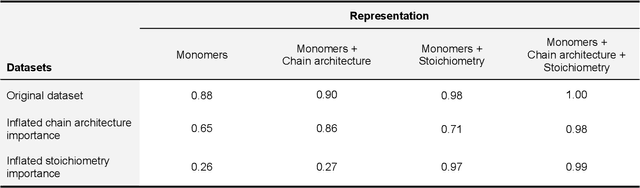
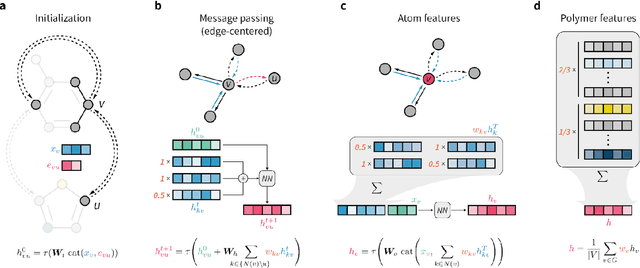

Synthetic polymers are versatile and widely used materials. Similar to small organic molecules, a large chemical space of such materials is hypothetically accessible. Computational property prediction and virtual screening can accelerate polymer design by prioritizing candidates expected to have favorable properties. However, in contrast to organic molecules, polymers are often not well-defined single structures but an ensemble of similar molecules, which poses unique challenges to traditional chemical representations and machine learning approaches. Here, we introduce a graph representation of molecular ensembles and an associated graph neural network architecture that is tailored to polymer property prediction. We demonstrate that this approach captures critical features of polymeric materials, like chain architecture, monomer stoichiometry, and degree of polymerization, and achieves superior accuracy to off-the-shelf cheminformatics methodologies. While doing so, we built a dataset of simulated electron affinity and ionization potential values for >40k polymers with varying monomer composition, stoichiometry, and chain architecture, which may be used in the development of other tailored machine learning approaches. The dataset and machine learning models presented in this work pave the path toward new classes of algorithms for polymer informatics and, more broadly, introduce a framework for the modeling of molecular ensembles.