 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCalibration and generalizability of probabilistic models on low-data chemical datasets with DIONYSUS
Dec 06, 2022Deep learning models that leverage large datasets are often the state of the art for modelling molecular properties. When the datasets are smaller (< 2000 molecules), it is not clear that deep learning approaches are the right modelling tool. In this work we perform an extensive study of the calibration and generalizability of probabilistic machine learning models on small chemical datasets. Using different molecular representations and models, we analyse the quality of their predictions and uncertainties in a variety of tasks (binary, regression) and datasets. We also introduce two simulated experiments that evaluate their performance: (1) Bayesian optimization guided molecular design, (2) inference on out-of-distribution data via ablated cluster splits. We offer practical insights into model and feature choice for modelling small chemical datasets, a common scenario in new chemical experiments. We have packaged our analysis into the DIONYSUS repository, which is open sourced to aid in reproducibility and extension to new datasets.
Bayesian optimization with known experimental and design constraints for chemistry applications
Mar 29, 2022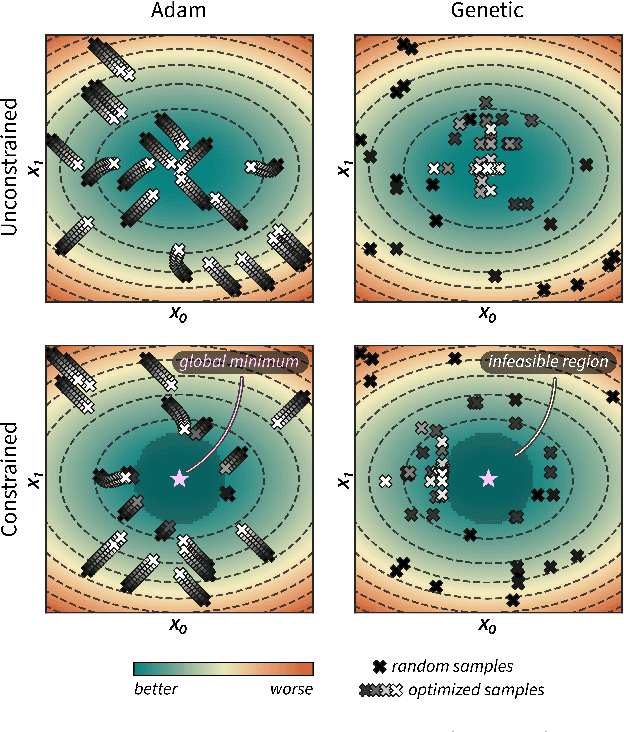
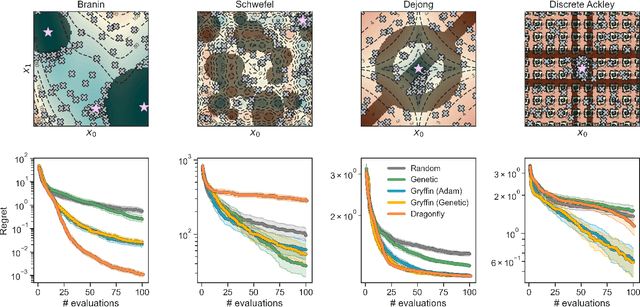

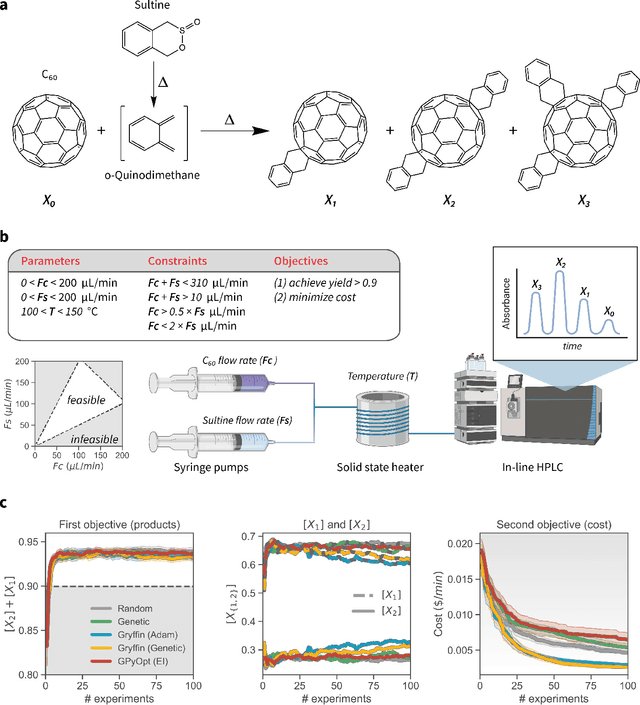
Optimization strategies driven by machine learning, such as Bayesian optimization, are being explored across experimental sciences as an efficient alternative to traditional design of experiment. When combined with automated laboratory hardware and high-performance computing, these strategies enable next-generation platforms for autonomous experimentation. However, the practical application of these approaches is hampered by a lack of flexible software and algorithms tailored to the unique requirements of chemical research. One such aspect is the pervasive presence of constraints in the experimental conditions when optimizing chemical processes or protocols, and in the chemical space that is accessible when designing functional molecules or materials. Although many of these constraints are known a priori, they can be interdependent, non-linear, and result in non-compact optimization domains. In this work, we extend our experiment planning algorithms Phoenics and Gryffin such that they can handle arbitrary known constraints via an intuitive and flexible interface. We benchmark these extended algorithms on continuous and discrete test functions with a diverse set of constraints, demonstrating their flexibility and robustness. In addition, we illustrate their practical utility in two simulated chemical research scenarios: the optimization of the synthesis of o-xylenyl Buckminsterfullerene adducts under constrained flow conditions, and the design of redox active molecules for flow batteries under synthetic accessibility constraints. The tools developed constitute a simple, yet versatile strategy to enable model-based optimization with known experimental constraints, contributing to its applicability as a core component of autonomous platforms for scientific discovery.
Golem: An algorithm for robust experiment and process optimization
Mar 05, 2021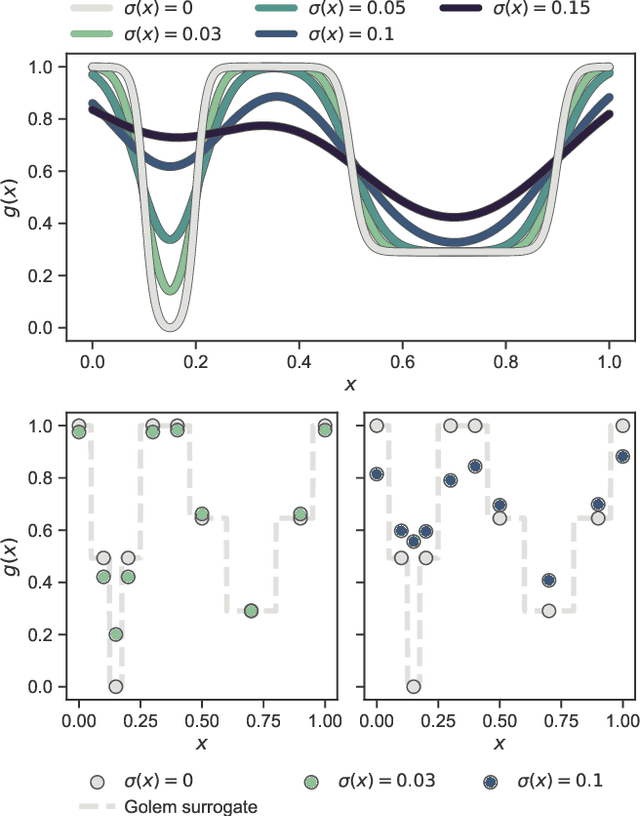
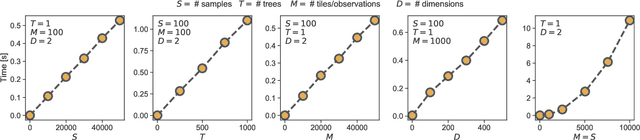
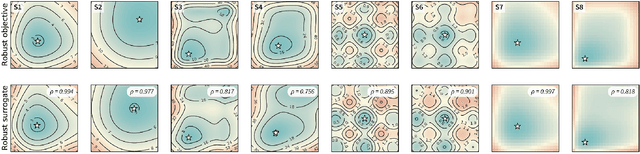
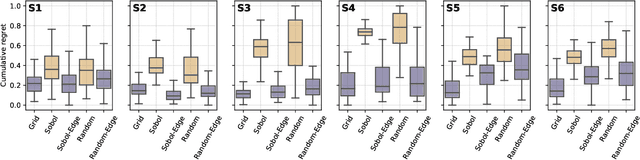
Numerous challenges in science and engineering can be framed as optimization tasks, including the maximization of reaction yields, the optimization of molecular and materials properties, and the fine-tuning of automated hardware protocols. Design of experiment and optimization algorithms are often adopted to solve these tasks efficiently. Increasingly, these experiment planning strategies are coupled with automated hardware to enable autonomous experimental platforms. The vast majority of the strategies used, however, do not consider robustness against the variability of experiment and process conditions. In fact, it is generally assumed that these parameters are exact and reproducible. Yet some experiments may have considerable noise associated with some of their conditions, and process parameters optimized under precise control may be applied in the future under variable operating conditions. In either scenario, the optimal solutions found might not be robust against input variability, affecting the reproducibility of results and returning suboptimal performance in practice. Here, we introduce Golem, an algorithm that is agnostic to the choice of experiment planning strategy and that enables robust experiment and process optimization. Golem identifies optimal solutions that are robust to input uncertainty, thus ensuring the reproducible performance of optimized experimental protocols and processes. It can be used to analyze the robustness of past experiments, or to guide experiment planning algorithms toward robust solutions on the fly. We assess the performance and domain of applicability of Golem through extensive benchmark studies and demonstrate its practical relevance by optimizing an analytical chemistry protocol under the presence of significant noise in its experimental conditions.
Gemini: Dynamic Bias Correction for Autonomous Experimentation and Molecular Simulation
Mar 05, 2021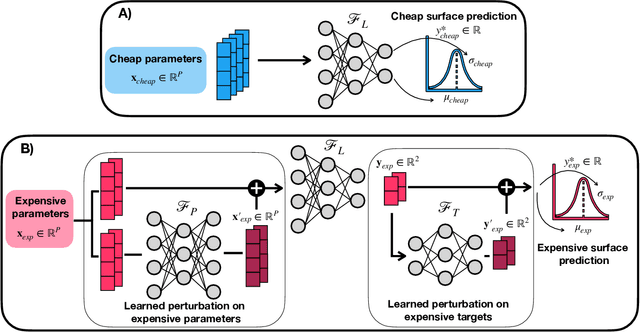
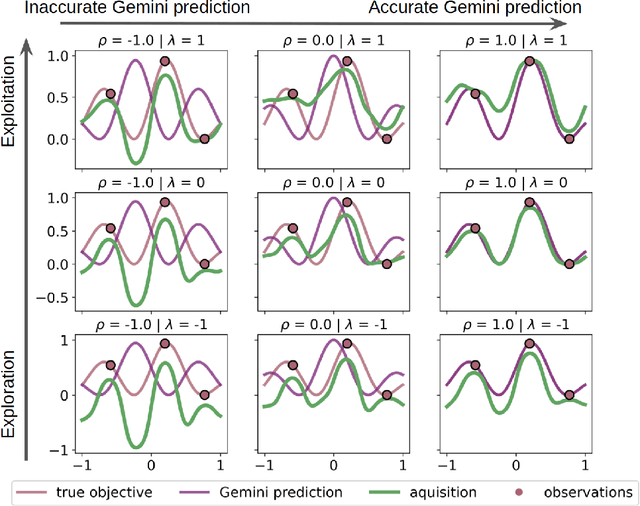
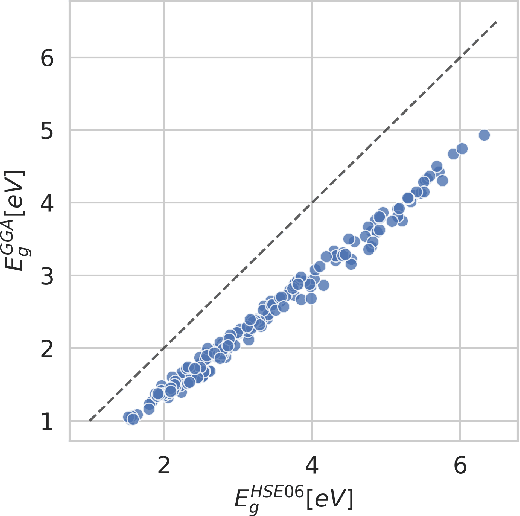
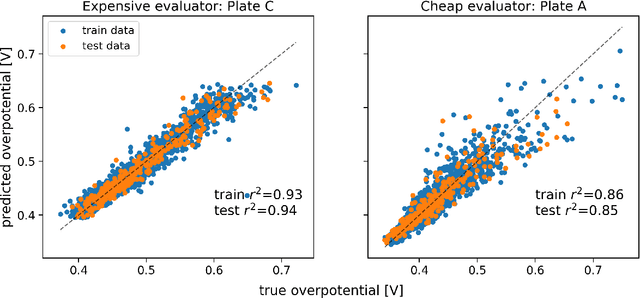
Bayesian optimization has emerged as a powerful strategy to accelerate scientific discovery by means of autonomous experimentation. However, expensive measurements are required to accurately estimate materials properties, and can quickly become a hindrance to exhaustive materials discovery campaigns. Here, we introduce Gemini: a data-driven model capable of using inexpensive measurements as proxies for expensive measurements by correcting systematic biases between property evaluation methods. We recommend using Gemini for regression tasks with sparse data and in an autonomous workflow setting where its predictions of expensive to evaluate objectives can be used to construct a more informative acquisition function, thus reducing the number of expensive evaluations an optimizer needs to achieve desired target values. In a regression setting, we showcase the ability of our method to make accurate predictions of DFT calculated bandgaps of hybrid organic-inorganic perovskite materials. We further demonstrate the benefits that Gemini provides to autonomous workflows by augmenting the Bayesian optimizer Phoenics to yeild a scalable optimization framework leveraging multiple sources of measurement. Finally, we simulate an autonomous materials discovery platform for optimizing the activity of electrocatalysts for the oxygen evolution reaction. Realizing autonomous workflows with Gemini, we show that the number of measurements of a composition space comprising expensive and rare metals needed to achieve a target overpotential is significantly reduced when measurements from a proxy composition system with less expensive metals are available.
Assigning Confidence to Molecular Property Prediction
Feb 23, 2021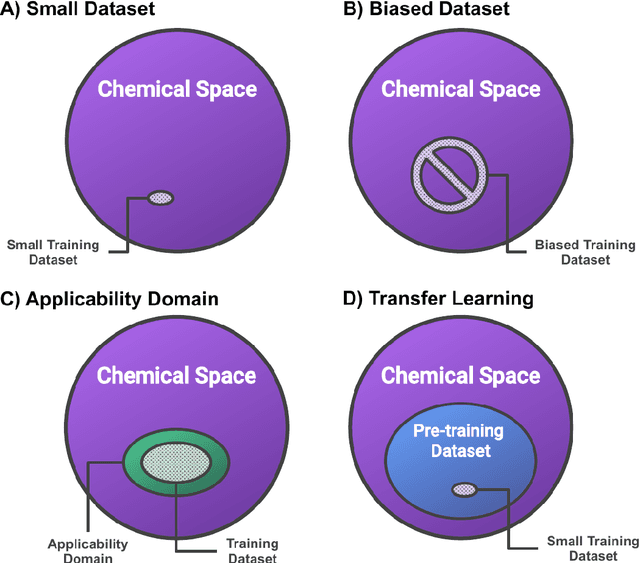
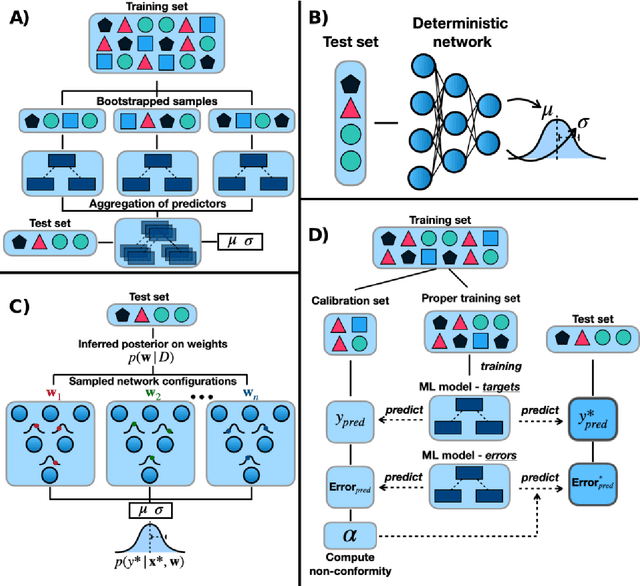
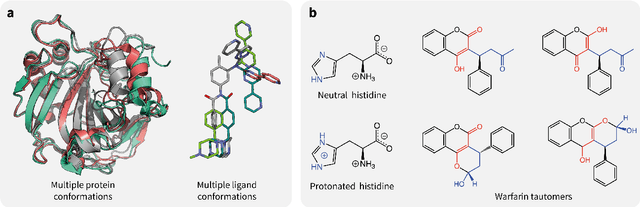
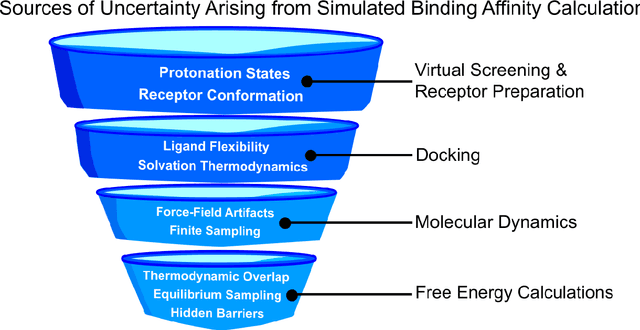
Introduction: Computational modeling has rapidly advanced over the last decades, especially to predict molecular properties for chemistry, material science and drug design. Recently, machine learning techniques have emerged as a powerful and cost-effective strategy to learn from existing datasets and perform predictions on unseen molecules. Accordingly, the explosive rise of data-driven techniques raises an important question: What confidence can be assigned to molecular property predictions and what techniques can be used for that purpose? Areas covered: In this work, we discuss popular strategies for predicting molecular properties relevant to drug design, their corresponding uncertainty sources and methods to quantify uncertainty and confidence. First, our considerations for assessing confidence begin with dataset bias and size, data-driven property prediction and feature design. Next, we discuss property simulation via molecular docking, and free-energy simulations of binding affinity in detail. Lastly, we investigate how these uncertainties propagate to generative models, as they are usually coupled with property predictors. Expert opinion: Computational techniques are paramount to reduce the prohibitive cost and timing of brute-force experimentation when exploring the enormous chemical space. We believe that assessing uncertainty in property prediction models is essential whenever closed-loop drug design campaigns relying on high-throughput virtual screening are deployed. Accordingly, considering sources of uncertainty leads to better-informed experimental validations, more reliable predictions and to more realistic expectations of the entire workflow. Overall, this increases confidence in the predictions and designs and, ultimately, accelerates drug design.
Olympus: a benchmarking framework for noisy optimization and experiment planning
Oct 08, 2020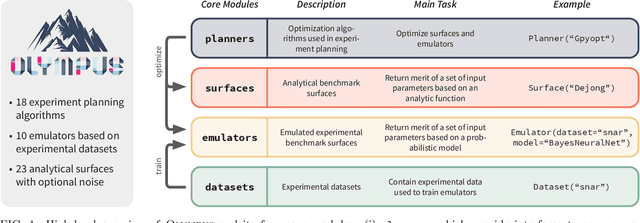
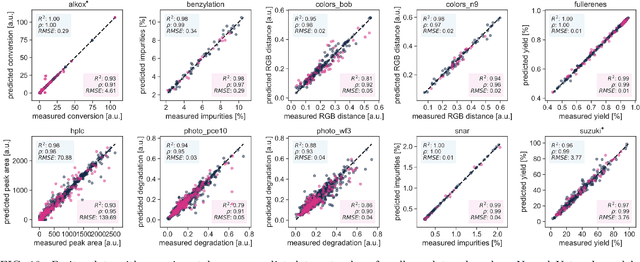

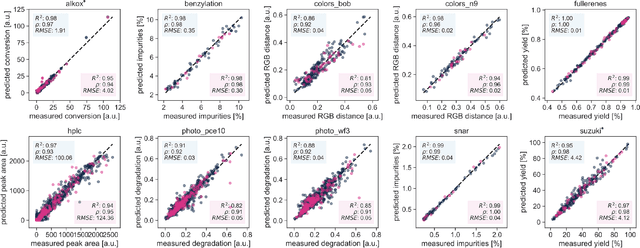
Research challenges encountered across science, engineering, and economics can frequently be formulated as optimization tasks. In chemistry and materials science, recent growth in laboratory digitization and automation has sparked interest in optimization-guided autonomous discovery and closed-loop experimentation. Experiment planning strategies based on off-the-shelf optimization algorithms can be employed in fully autonomous research platforms to achieve desired experimentation goals with the minimum number of trials. However, the experiment planning strategy that is most suitable to a scientific discovery task is a priori unknown while rigorous comparisons of different strategies are highly time and resource demanding. As optimization algorithms are typically benchmarked on low-dimensional synthetic functions, it is unclear how their performance would translate to noisy, higher-dimensional experimental tasks encountered in chemistry and materials science. We introduce Olympus, a software package that provides a consistent and easy-to-use framework for benchmarking optimization algorithms against realistic experiments emulated via probabilistic deep-learning models. Olympus includes a collection of experimentally derived benchmark sets from chemistry and materials science and a suite of experiment planning strategies that can be easily accessed via a user-friendly python interface. Furthermore, Olympus facilitates the integration, testing, and sharing of custom algorithms and user-defined datasets. In brief, Olympus mitigates the barriers associated with benchmarking optimization algorithms on realistic experimental scenarios, promoting data sharing and the creation of a standard framework for evaluating the performance of experiment planning strategies