 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMATTERIX: toward a digital twin for robotics-assisted chemistry laboratory automation
Jan 19, 2026Accelerated materials discovery is critical for addressing global challenges. However, developing new laboratory workflows relies heavily on real-world experimental trials, and this can hinder scalability because of the need for numerous physical make-and-test iterations. Here we present MATTERIX, a multiscale, graphics processing unit-accelerated robotic simulation framework designed to create high-fidelity digital twins of chemistry laboratories, thus accelerating workflow development. This multiscale digital twin simulates robotic physical manipulation, powder and liquid dynamics, device functionalities, heat transfer and basic chemical reaction kinetics. This is enabled by integrating realistic physics simulation and photorealistic rendering with a modular graphics processing unit-accelerated semantics engine, which models logical states and continuous behaviors to simulate chemistry workflows across different levels of abstraction. MATTERIX streamlines the creation of digital twin environments through open-source asset libraries and interfaces, while enabling flexible workflow design via hierarchical plan definition and a modular skill library that incorporates learning-based methods. Our approach demonstrates sim-to-real transfer in robotic chemistry setups, reducing reliance on costly real-world experiments and enabling the testing of hypothetical automated workflows in silico. The project website is available at https://accelerationconsortium.github.io/Matterix/ .
El Agente: An Autonomous Agent for Quantum Chemistry
May 05, 2025Computational chemistry tools are widely used to study the behaviour of chemical phenomena. Yet, the complexity of these tools can make them inaccessible to non-specialists and challenging even for experts. In this work, we introduce El Agente Q, an LLM-based multi-agent system that dynamically generates and executes quantum chemistry workflows from natural language user prompts. The system is built on a novel cognitive architecture featuring a hierarchical memory framework that enables flexible task decomposition, adaptive tool selection, post-analysis, and autonomous file handling and submission. El Agente Q is benchmarked on six university-level course exercises and two case studies, demonstrating robust problem-solving performance (averaging >87% task success) and adaptive error handling through in situ debugging. It also supports longer-term, multi-step task execution for more complex workflows, while maintaining transparency through detailed action trace logs. Together, these capabilities lay the foundation for increasingly autonomous and accessible quantum chemistry.
From Molecules to Mixtures: Learning Representations of Olfactory Mixture Similarity using Inductive Biases
Jan 27, 2025



Olfaction -- how molecules are perceived as odors to humans -- remains poorly understood. Recently, the principal odor map (POM) was introduced to digitize the olfactory properties of single compounds. However, smells in real life are not pure single molecules, but complex mixtures of molecules, whose representations remain relatively under-explored. In this work, we introduce POMMix, an extension of the POM to represent mixtures. Our representation builds upon the symmetries of the problem space in a hierarchical manner: (1) graph neural networks for building molecular embeddings, (2) attention mechanisms for aggregating molecular representations into mixture representations, and (3) cosine prediction heads to encode olfactory perceptual distance in the mixture embedding space. POMMix achieves state-of-the-art predictive performance across multiple datasets. We also evaluate the generalizability of the representation on multiple splits when applied to unseen molecules and mixture sizes. Our work advances the effort to digitize olfaction, and highlights the synergy of domain expertise and deep learning in crafting expressive representations in low-data regimes.
Ranking over Regression for Bayesian Optimization and Molecule Selection
Oct 11, 2024



Bayesian optimization (BO) has become an indispensable tool for autonomous decision-making across diverse applications from autonomous vehicle control to accelerated drug and materials discovery. With the growing interest in self-driving laboratories, BO of chemical systems is crucial for machine learning (ML) guided experimental planning. Typically, BO employs a regression surrogate model to predict the distribution of unseen parts of the search space. However, for the selection of molecules, picking the top candidates with respect to a distribution, the relative ordering of their properties may be more important than their exact values. In this paper, we introduce Rank-based Bayesian Optimization (RBO), which utilizes a ranking model as the surrogate. We present a comprehensive investigation of RBO's optimization performance compared to conventional BO on various chemical datasets. Our results demonstrate similar or improved optimization performance using ranking models, particularly for datasets with rough structure-property landscapes and activity cliffs. Furthermore, we observe a high correlation between the surrogate ranking ability and BO performance, and this ability is maintained even at early iterations of BO optimization when using ranking surrogate models. We conclude that RBO is an effective alternative to regression-based BO, especially for optimizing novel chemical compounds.
Calibration and generalizability of probabilistic models on low-data chemical datasets with DIONYSUS
Dec 06, 2022Deep learning models that leverage large datasets are often the state of the art for modelling molecular properties. When the datasets are smaller (< 2000 molecules), it is not clear that deep learning approaches are the right modelling tool. In this work we perform an extensive study of the calibration and generalizability of probabilistic machine learning models on small chemical datasets. Using different molecular representations and models, we analyse the quality of their predictions and uncertainties in a variety of tasks (binary, regression) and datasets. We also introduce two simulated experiments that evaluate their performance: (1) Bayesian optimization guided molecular design, (2) inference on out-of-distribution data via ablated cluster splits. We offer practical insights into model and feature choice for modelling small chemical datasets, a common scenario in new chemical experiments. We have packaged our analysis into the DIONYSUS repository, which is open sourced to aid in reproducibility and extension to new datasets.
SELFIES and the future of molecular string representations
Mar 31, 2022
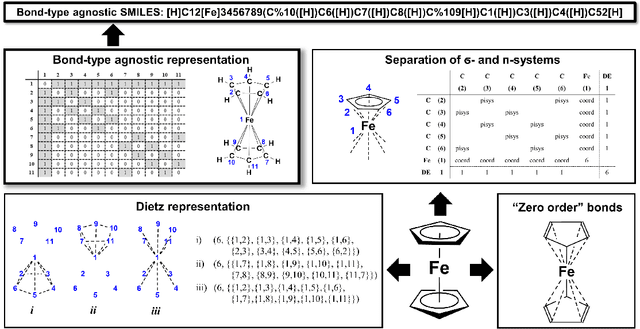
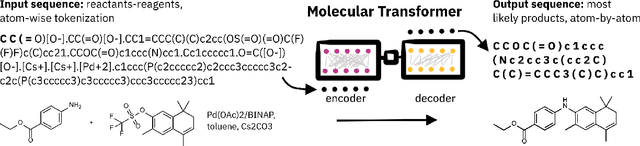
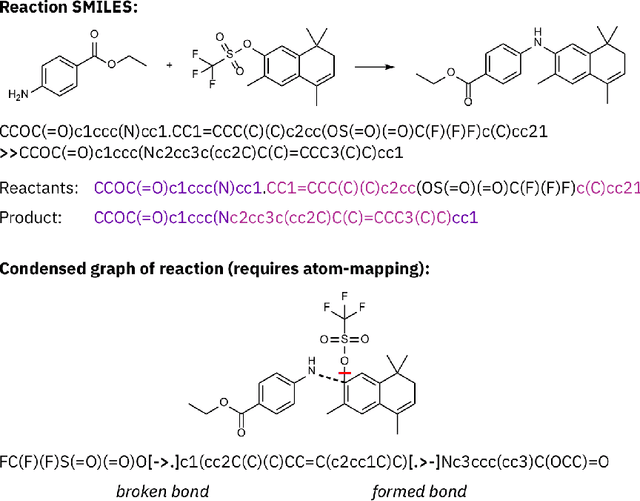
Artificial intelligence (AI) and machine learning (ML) are expanding in popularity for broad applications to challenging tasks in chemistry and materials science. Examples include the prediction of properties, the discovery of new reaction pathways, or the design of new molecules. The machine needs to read and write fluently in a chemical language for each of these tasks. Strings are a common tool to represent molecular graphs, and the most popular molecular string representation, SMILES, has powered cheminformatics since the late 1980s. However, in the context of AI and ML in chemistry, SMILES has several shortcomings -- most pertinently, most combinations of symbols lead to invalid results with no valid chemical interpretation. To overcome this issue, a new language for molecules was introduced in 2020 that guarantees 100\% robustness: SELFIES (SELF-referencIng Embedded Strings). SELFIES has since simplified and enabled numerous new applications in chemistry. In this manuscript, we look to the future and discuss molecular string representations, along with their respective opportunities and challenges. We propose 16 concrete Future Projects for robust molecular representations. These involve the extension toward new chemical domains, exciting questions at the interface of AI and robust languages and interpretability for both humans and machines. We hope that these proposals will inspire several follow-up works exploiting the full potential of molecular string representations for the future of AI in chemistry and materials science.