 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGraph Data Modeling: Molecules, Proteins, & Chemical Processes
Aug 28, 2025Graphs are central to the chemical sciences, providing a natural language to describe molecules, proteins, reactions, and industrial processes. They capture interactions and structures that underpin materials, biology, and medicine. This primer, Graph Data Modeling: Molecules, Proteins, & Chemical Processes, introduces graphs as mathematical objects in chemistry and shows how learning algorithms (particularly graph neural networks) can operate on them. We outline the foundations of graph design, key prediction tasks, representative examples across chemical sciences, and the role of machine learning in graph-based modeling. Together, these concepts prepare readers to apply graph methods to the next generation of chemical discovery.
CheMixHub: Datasets and Benchmarks for Chemical Mixture Property Prediction
Jun 13, 2025Developing improved predictive models for multi-molecular systems is crucial, as nearly every chemical product used results from a mixture of chemicals. While being a vital part of the industry pipeline, the chemical mixture space remains relatively unexplored by the Machine Learning community. In this paper, we introduce CheMixHub, a holistic benchmark for molecular mixtures, covering a corpus of 11 chemical mixtures property prediction tasks, from drug delivery formulations to battery electrolytes, totalling approximately 500k data points gathered and curated from 7 publicly available datasets. CheMixHub introduces various data splitting techniques to assess context-specific generalization and model robustness, providing a foundation for the development of predictive models for chemical mixture properties. Furthermore, we map out the modelling space of deep learning models for chemical mixtures, establishing initial benchmarks for the community. This dataset has the potential to accelerate chemical mixture development, encompassing reformulation, optimization, and discovery. The dataset and code for the benchmarks can be found at: https://github.com/chemcognition-lab/chemixhub
From Molecules to Mixtures: Learning Representations of Olfactory Mixture Similarity using Inductive Biases
Jan 27, 2025



Olfaction -- how molecules are perceived as odors to humans -- remains poorly understood. Recently, the principal odor map (POM) was introduced to digitize the olfactory properties of single compounds. However, smells in real life are not pure single molecules, but complex mixtures of molecules, whose representations remain relatively under-explored. In this work, we introduce POMMix, an extension of the POM to represent mixtures. Our representation builds upon the symmetries of the problem space in a hierarchical manner: (1) graph neural networks for building molecular embeddings, (2) attention mechanisms for aggregating molecular representations into mixture representations, and (3) cosine prediction heads to encode olfactory perceptual distance in the mixture embedding space. POMMix achieves state-of-the-art predictive performance across multiple datasets. We also evaluate the generalizability of the representation on multiple splits when applied to unseen molecules and mixture sizes. Our work advances the effort to digitize olfaction, and highlights the synergy of domain expertise and deep learning in crafting expressive representations in low-data regimes.
Ranking over Regression for Bayesian Optimization and Molecule Selection
Oct 11, 2024



Bayesian optimization (BO) has become an indispensable tool for autonomous decision-making across diverse applications from autonomous vehicle control to accelerated drug and materials discovery. With the growing interest in self-driving laboratories, BO of chemical systems is crucial for machine learning (ML) guided experimental planning. Typically, BO employs a regression surrogate model to predict the distribution of unseen parts of the search space. However, for the selection of molecules, picking the top candidates with respect to a distribution, the relative ordering of their properties may be more important than their exact values. In this paper, we introduce Rank-based Bayesian Optimization (RBO), which utilizes a ranking model as the surrogate. We present a comprehensive investigation of RBO's optimization performance compared to conventional BO on various chemical datasets. Our results demonstrate similar or improved optimization performance using ranking models, particularly for datasets with rough structure-property landscapes and activity cliffs. Furthermore, we observe a high correlation between the surrogate ranking ability and BO performance, and this ability is maintained even at early iterations of BO optimization when using ranking surrogate models. We conclude that RBO is an effective alternative to regression-based BO, especially for optimizing novel chemical compounds.
Advancing Molecular Machine (Learned) Representations with Stereoelectronics-Infused Molecular Graphs
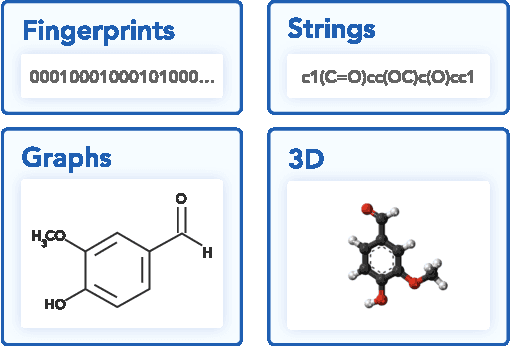
Aug 08, 2024Molecular representation is a foundational element in our understanding of the physical world. Its importance ranges from the fundamentals of chemical reactions to the design of new therapies and materials. Previous molecular machine learning models have employed strings, fingerprints, global features, and simple molecular graphs that are inherently information-sparse representations. However, as the complexity of prediction tasks increases, the molecular representation needs to encode higher fidelity information. This work introduces a novel approach to infusing quantum-chemical-rich information into molecular graphs via stereoelectronic effects. We show that the explicit addition of stereoelectronic interactions significantly improves the performance of molecular machine learning models. Furthermore, stereoelectronics-infused representations can be learned and deployed with a tailored double graph neural network workflow, enabling its application to any downstream molecular machine learning task. Finally, we show that the learned representations allow for facile stereoelectronic evaluation of previously intractable systems, such as entire proteins, opening new avenues of molecular design.
Calibration and generalizability of probabilistic models on low-data chemical datasets with DIONYSUS
Dec 06, 2022Deep learning models that leverage large datasets are often the state of the art for modelling molecular properties. When the datasets are smaller (< 2000 molecules), it is not clear that deep learning approaches are the right modelling tool. In this work we perform an extensive study of the calibration and generalizability of probabilistic machine learning models on small chemical datasets. Using different molecular representations and models, we analyse the quality of their predictions and uncertainties in a variety of tasks (binary, regression) and datasets. We also introduce two simulated experiments that evaluate their performance: (1) Bayesian optimization guided molecular design, (2) inference on out-of-distribution data via ablated cluster splits. We offer practical insights into model and feature choice for modelling small chemical datasets, a common scenario in new chemical experiments. We have packaged our analysis into the DIONYSUS repository, which is open sourced to aid in reproducibility and extension to new datasets.
Machine Learning for Scent: Learning Generalizable Perceptual Representations of Small Molecules
Oct 25, 2019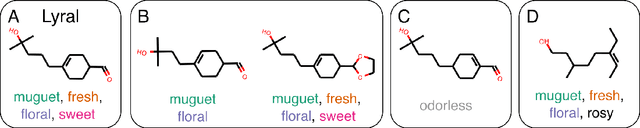
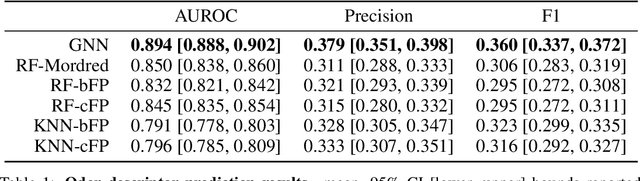
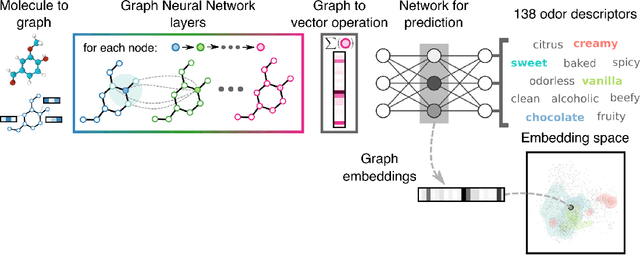
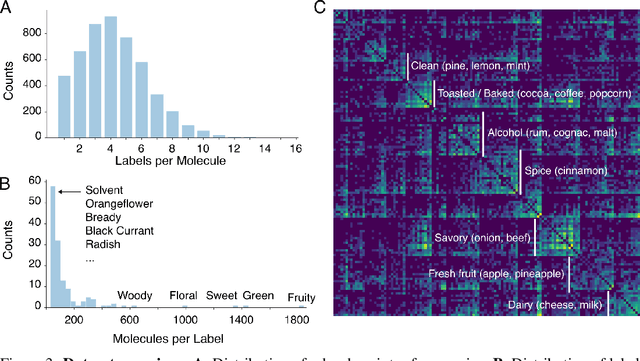
Predicting the relationship between a molecule's structure and its odor remains a difficult, decades-old task. This problem, termed quantitative structure-odor relationship (QSOR) modeling, is an important challenge in chemistry, impacting human nutrition, manufacture of synthetic fragrance, the environment, and sensory neuroscience. We propose the use of graph neural networks for QSOR, and show they significantly out-perform prior methods on a novel data set labeled by olfactory experts. Additional analysis shows that the learned embeddings from graph neural networks capture a meaningful odor space representation of the underlying relationship between structure and odor, as demonstrated by strong performance on two challenging transfer learning tasks. Machine learning has already had a large impact on the senses of sight and sound. Based on these early results with graph neural networks for molecular properties, we hope machine learning can eventually do for olfaction what it has already done for vision and hearing.
Molecular Sets (MOSES): A Benchmarking Platform for Molecular Generation Models
Nov 29, 2018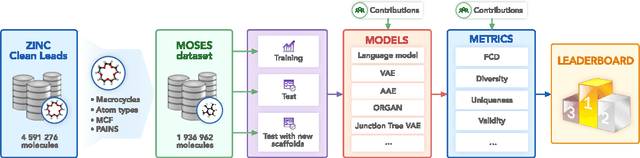


Deep generative models such as generative adversarial networks, variational autoencoders, and autoregressive models are rapidly growing in popularity for the discovery of new molecules and materials. In this work, we introduce MOlecular SEtS (MOSES), a benchmarking platform to support research on machine learning for drug discovery. MOSES implements several popular molecular generation models and includes a set of metrics that evaluate the diversity and quality of generated molecules. MOSES is meant to standardize the research on the molecular generation and facilitate the sharing and comparison of new models. Additionally, we provide a large-scale comparison of existing state of the art models and elaborate on current challenges for generative models that might prove fertile ground for new research. Our platform and source code are freely available at https://github.com/molecularsets/
Objective-Reinforced Generative Adversarial Networks (ORGAN) for Sequence Generation Models
Feb 07, 2018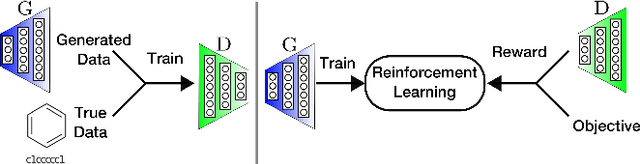



In unsupervised data generation tasks, besides the generation of a sample based on previous observations, one would often like to give hints to the model in order to bias the generation towards desirable metrics. We propose a method that combines Generative Adversarial Networks (GANs) and reinforcement learning (RL) in order to accomplish exactly that. While RL biases the data generation process towards arbitrary metrics, the GAN component of the reward function ensures that the model still remembers information learned from data. We build upon previous results that incorporated GANs and RL in order to generate sequence data and test this model in several settings for the generation of molecules encoded as text sequences (SMILES) and in the context of music generation, showing for each case that we can effectively bias the generation process towards desired metrics.