 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMMAI Gym for Science: Training Liquid Foundation Models for Drug Discovery
Mar 03, 2026General-purpose large language models (LLMs) that rely on in-context learning do not reliably deliver the scientific understanding and performance required for drug discovery tasks. Simply increasing model size or introducing reasoning tokens does not yield significant performance gains. To address this gap, we introduce the MMAI Gym for Science, a one-stop shop molecular data formats and modalities as well as task-specific reasoning, training, and benchmarking recipes designed to teach foundation models the 'language of molecules' in order to solve practical drug discovery problems. We use MMAI Gym to train an efficient Liquid Foundation Model (LFM) for these applications, demonstrating that smaller, purpose-trained foundation models can outperform substantially larger general-purpose or specialist models on molecular benchmarks. Across essential drug discovery tasks - including molecular optimization, ADMET property prediction, retrosynthesis, drug-target activity prediction, and functional group reasoning - the resulting model achieves near specialist-level performance and, in the majority of settings, surpasses larger models, while remaining more efficient and broadly applicable in the domain.
When Single Answer Is Not Enough: Rethinking Single-Step Retrosynthesis Benchmarks for LLMs
Feb 03, 2026Recent progress has expanded the use of large language models (LLMs) in drug discovery, including synthesis planning. However, objective evaluation of retrosynthesis performance remains limited. Existing benchmarks and metrics typically rely on published synthetic procedures and Top-K accuracy based on single ground-truth, which does not capture the open-ended nature of real-world synthesis planning. We propose a new benchmarking framework for single-step retrosynthesis that evaluates both general-purpose and chemistry-specialized LLMs using ChemCensor, a novel metric for chemical plausibility. By emphasizing plausibility over exact match, this approach better aligns with human synthesis planning practices. We also introduce CREED, a novel dataset comprising millions of ChemCensor-validated reaction records for LLM training, and use it to train a model that improves over the LLM baselines under this benchmark.
AlphaFold Accelerates Artificial Intelligence Powered Drug Discovery: Efficient Discovery of a Novel Cyclin-dependent Kinase 20 (CDK20) Small Molecule Inhibitor
Jan 21, 2022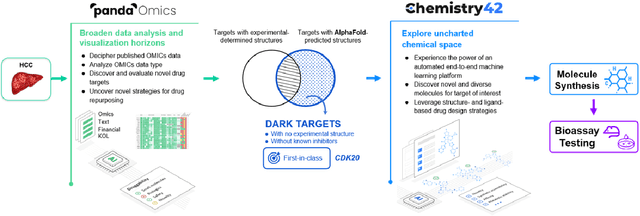
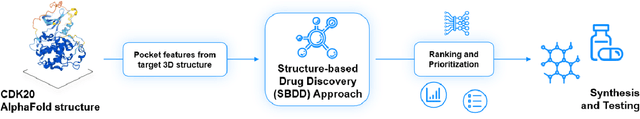
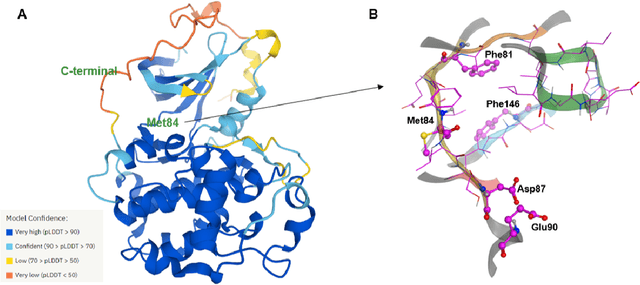
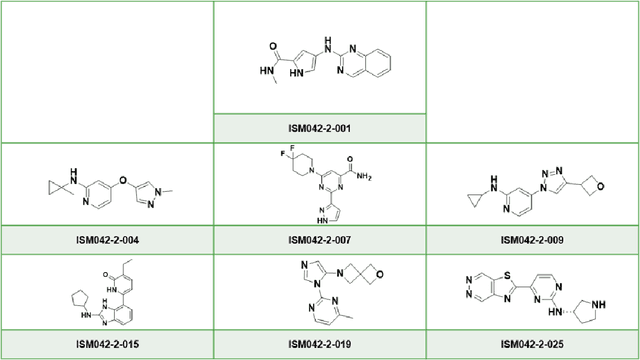
The AlphaFold computer program predicted protein structures for the whole human genome, which has been considered as a remarkable breakthrough both in artificial intelligence (AI) application and structural biology. Despite the varying confidence level, these predicted structures still could significantly contribute to the structure-based drug design of novel targets, especially the ones with no or limited structural information. In this work, we successfully applied AlphaFold in our end-to-end AI-powered drug discovery engines constituted of a biocomputational platform PandaOmics and a generative chemistry platform Chemistry42, to identify a first-in-class hit molecule of a novel target without an experimental structure starting from target selection towards hit identification in a cost- and time-efficient manner. PandaOmics provided the targets of interest and Chemistry42 generated the molecules based on the AlphaFold predicted structure, and the selected molecules were synthesized and tested in biological assays. Through this approach, we identified a small molecule hit compound for CDK20 with a Kd value of 8.9 +/- 1.6 uM (n = 4) within 30 days from target selection and after only synthesizing 7 compounds. To the best of our knowledge, this is the first reported small molecule targeting CDK20 and more importantly, this work is the first demonstration of AlphaFold application in the hit identification process in early drug discovery.
Chemistry42: An AI-based platform for de novo molecular design
Jan 22, 2021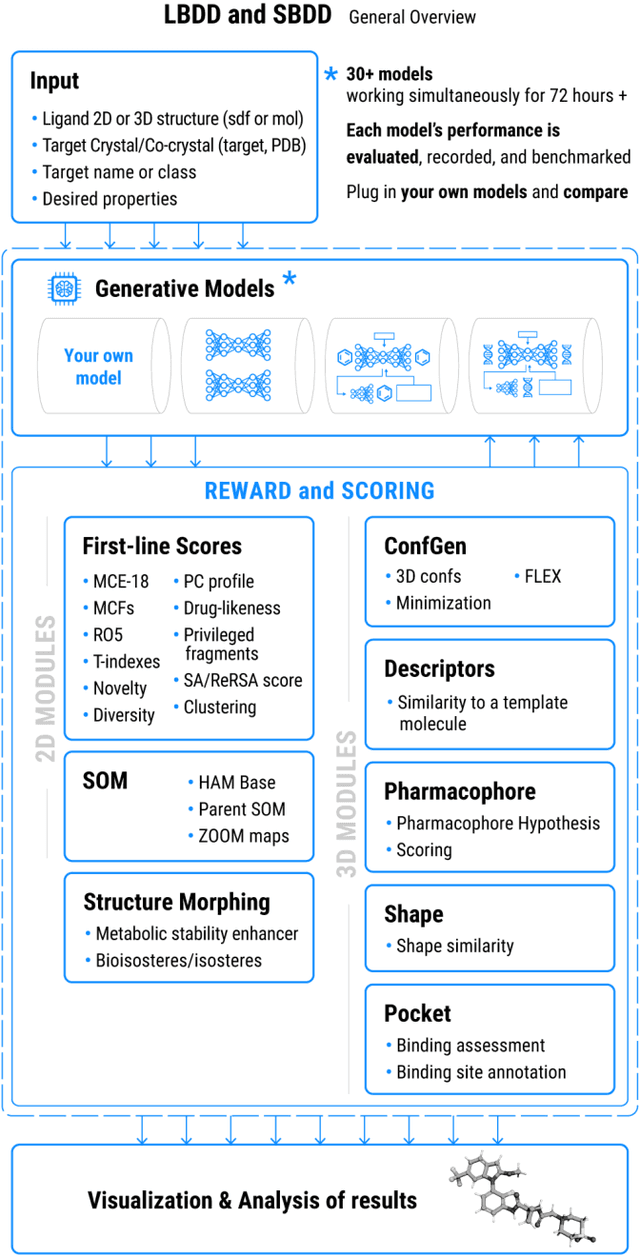
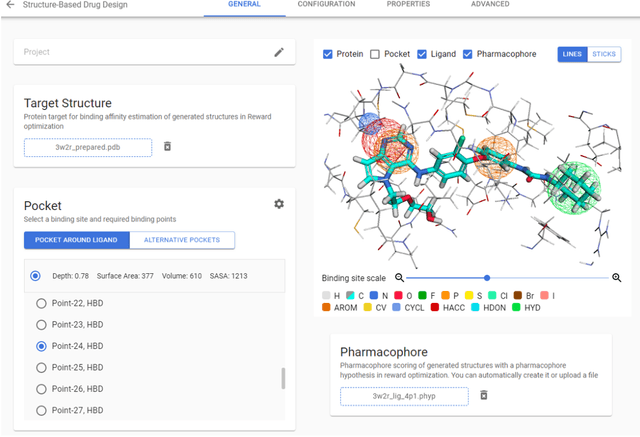
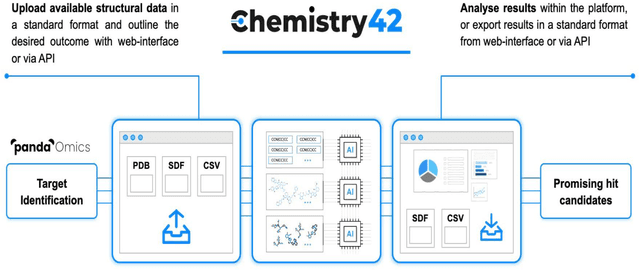
Chemistry42 is a software platform for de novo small molecule design that integrates Artificial Intelligence (AI) techniques with computational and medicinal chemistry methods. Chemistry42 is unique in its ability to generate novel molecular structures with predefined properties validated through in vitro and in vivo studies. Chemistry42 is a core component of Insilico Medicine Pharma.ai drug discovery suite that also includes target discovery and multi-omics data analysis (PandaOmics) and clinical trial outcomes predictions (InClinico).
Molecular Sets (MOSES): A Benchmarking Platform for Molecular Generation Models
Nov 29, 2018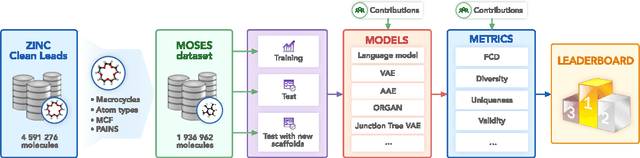


Deep generative models such as generative adversarial networks, variational autoencoders, and autoregressive models are rapidly growing in popularity for the discovery of new molecules and materials. In this work, we introduce MOlecular SEtS (MOSES), a benchmarking platform to support research on machine learning for drug discovery. MOSES implements several popular molecular generation models and includes a set of metrics that evaluate the diversity and quality of generated molecules. MOSES is meant to standardize the research on the molecular generation and facilitate the sharing and comparison of new models. Additionally, we provide a large-scale comparison of existing state of the art models and elaborate on current challenges for generative models that might prove fertile ground for new research. Our platform and source code are freely available at https://github.com/molecularsets/