 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeWhen Single Answer Is Not Enough: Rethinking Single-Step Retrosynthesis Benchmarks for LLMs
Feb 03, 2026Recent progress has expanded the use of large language models (LLMs) in drug discovery, including synthesis planning. However, objective evaluation of retrosynthesis performance remains limited. Existing benchmarks and metrics typically rely on published synthetic procedures and Top-K accuracy based on single ground-truth, which does not capture the open-ended nature of real-world synthesis planning. We propose a new benchmarking framework for single-step retrosynthesis that evaluates both general-purpose and chemistry-specialized LLMs using ChemCensor, a novel metric for chemical plausibility. By emphasizing plausibility over exact match, this approach better aligns with human synthesis planning practices. We also introduce CREED, a novel dataset comprising millions of ChemCensor-validated reaction records for LLM training, and use it to train a model that improves over the LLM baselines under this benchmark.
nach0-pc: Multi-task Language Model with Molecular Point Cloud Encoder
Oct 11, 2024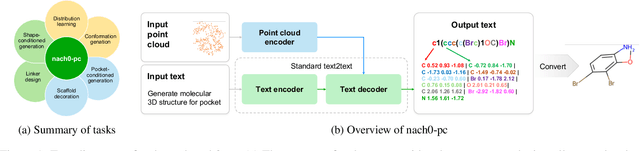
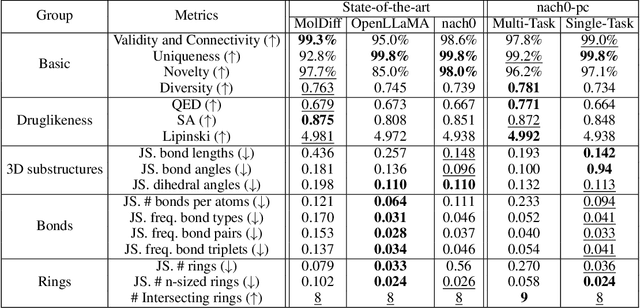
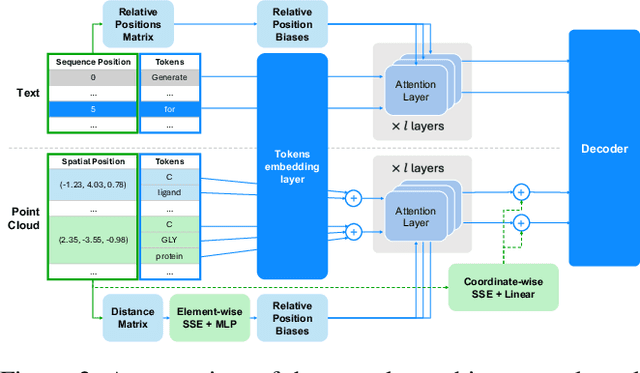
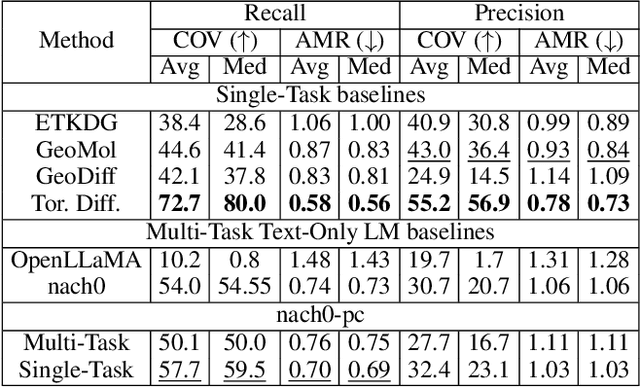
Recent advancements have integrated Language Models (LMs) into a drug discovery pipeline. However, existing models mostly work with SMILES and SELFIES chemical string representations, which lack spatial features vital for drug discovery. Additionally, attempts to translate chemical 3D structures into text format encounter issues such as excessive length and insufficient atom connectivity information. To address these issues, we introduce nach0-pc, a model combining domain-specific encoder and textual representation to handle spatial arrangement of atoms effectively. Our approach utilizes a molecular point cloud encoder for concise and order-invariant structure representation. We introduce a novel pre-training scheme for molecular point clouds to distillate the knowledge from spatial molecular structures datasets. After fine-tuning within both single-task and multi-task frameworks, nach0-pc demonstrates performance comparable with other diffusion models in terms of generated samples quality across several established spatial molecular generation tasks. Notably, our model is a multi-task approach, in contrast to diffusion models being limited to single tasks. Additionally, it is capable of processing point cloud-related data, which language models are not capable of handling due to memory limitations. These lead to our model having reduced training and inference time while maintaining on par performance.
Quantum Computing-Enhanced Algorithm Unveils Novel Inhibitors for KRAS
Feb 13, 2024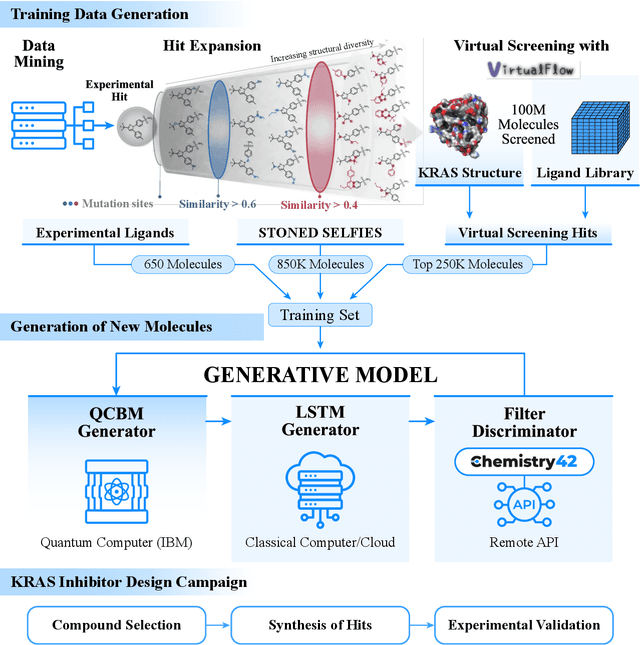
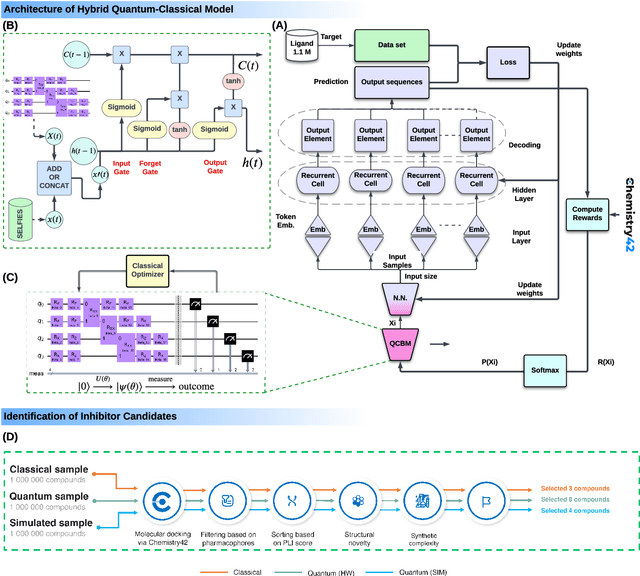
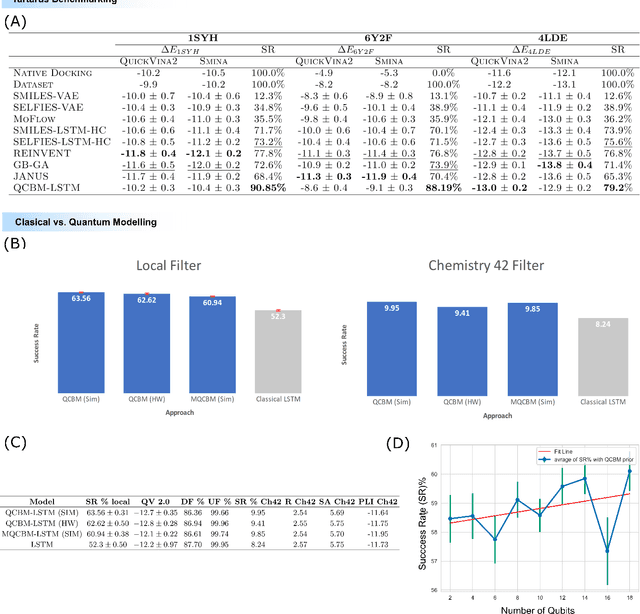
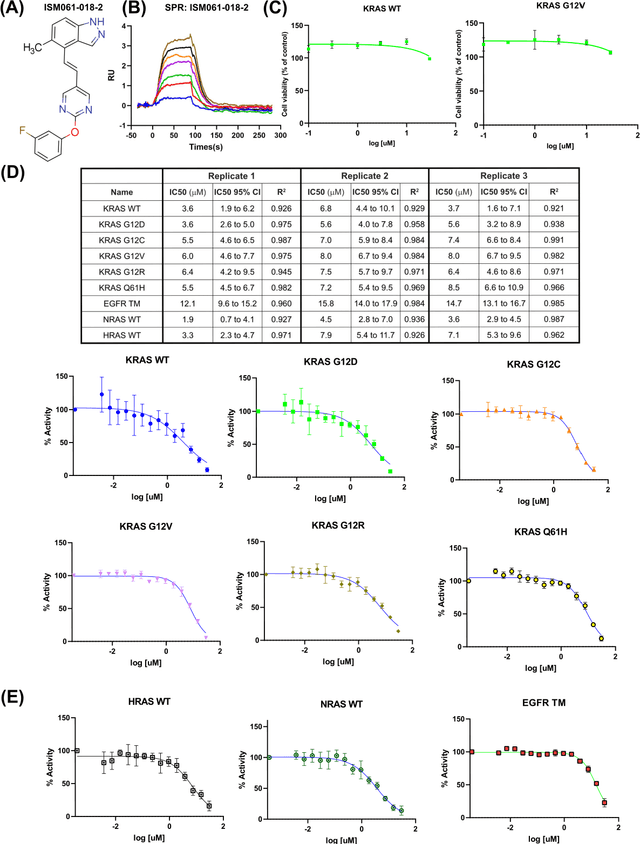
The discovery of small molecules with therapeutic potential is a long-standing challenge in chemistry and biology. Researchers have increasingly leveraged novel computational techniques to streamline the drug development process to increase hit rates and reduce the costs associated with bringing a drug to market. To this end, we introduce a quantum-classical generative model that seamlessly integrates the computational power of quantum algorithms trained on a 16-qubit IBM quantum computer with the established reliability of classical methods for designing small molecules. Our hybrid generative model was applied to designing new KRAS inhibitors, a crucial target in cancer therapy. We synthesized 15 promising molecules during our investigation and subjected them to experimental testing to assess their ability to engage with the target. Notably, among these candidates, two molecules, ISM061-018-2 and ISM061-22, each featuring unique scaffolds, stood out by demonstrating effective engagement with KRAS. ISM061-018-2 was identified as a broad-spectrum KRAS inhibitor, exhibiting a binding affinity to KRAS-G12D at $1.4 \mu M$. Concurrently, ISM061-22 exhibited specific mutant selectivity, displaying heightened activity against KRAS G12R and Q61H mutants. To our knowledge, this work shows for the first time the use of a quantum-generative model to yield experimentally confirmed biological hits, showcasing the practical potential of quantum-assisted drug discovery to produce viable therapeutics. Moreover, our findings reveal that the efficacy of distribution learning correlates with the number of qubits utilized, underlining the scalability potential of quantum computing resources. Overall, we anticipate our results to be a stepping stone towards developing more advanced quantum generative models in drug discovery.
nach0: Multimodal Natural and Chemical Languages Foundation Model
Nov 21, 2023



Large Language Models (LLMs) have substantially driven scientific progress in various domains, and many papers have demonstrated their ability to tackle complex problems with creative solutions. Our paper introduces a new foundation model, nach0, capable of solving various chemical and biological tasks: biomedical question answering, named entity recognition, molecular generation, molecular synthesis, attributes prediction, and others. nach0 is a multi-domain and multi-task encoder-decoder LLM pre-trained on unlabeled text from scientific literature, patents, and molecule strings to incorporate a range of chemical and linguistic knowledge. We employed instruction tuning, where specific task-related instructions are utilized to fine-tune nach0 for the final set of tasks. To train nach0 effectively, we leverage the NeMo framework, enabling efficient parallel optimization of both base and large model versions. Extensive experiments demonstrate that our model outperforms state-of-the-art baselines on single-domain and cross-domain tasks. Furthermore, it can generate high-quality outputs in molecular and textual formats, showcasing its effectiveness in multi-domain setups.
AlphaFold Accelerates Artificial Intelligence Powered Drug Discovery: Efficient Discovery of a Novel Cyclin-dependent Kinase 20 (CDK20) Small Molecule Inhibitor
Jan 21, 2022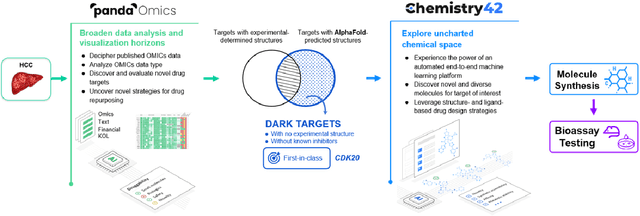
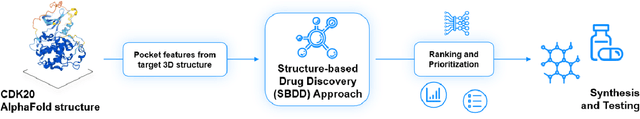
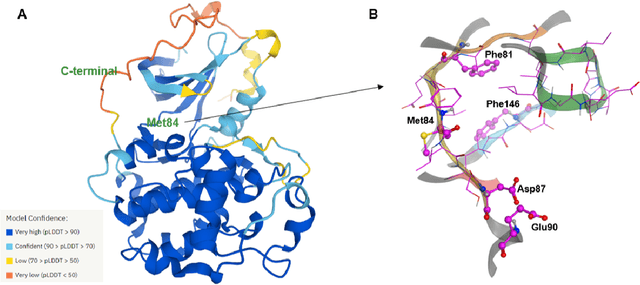
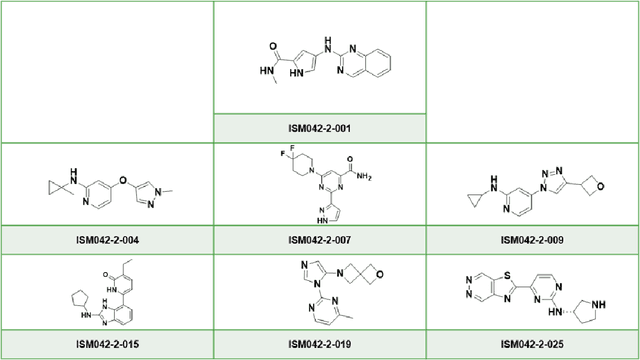
The AlphaFold computer program predicted protein structures for the whole human genome, which has been considered as a remarkable breakthrough both in artificial intelligence (AI) application and structural biology. Despite the varying confidence level, these predicted structures still could significantly contribute to the structure-based drug design of novel targets, especially the ones with no or limited structural information. In this work, we successfully applied AlphaFold in our end-to-end AI-powered drug discovery engines constituted of a biocomputational platform PandaOmics and a generative chemistry platform Chemistry42, to identify a first-in-class hit molecule of a novel target without an experimental structure starting from target selection towards hit identification in a cost- and time-efficient manner. PandaOmics provided the targets of interest and Chemistry42 generated the molecules based on the AlphaFold predicted structure, and the selected molecules were synthesized and tested in biological assays. Through this approach, we identified a small molecule hit compound for CDK20 with a Kd value of 8.9 +/- 1.6 uM (n = 4) within 30 days from target selection and after only synthesizing 7 compounds. To the best of our knowledge, this is the first reported small molecule targeting CDK20 and more importantly, this work is the first demonstration of AlphaFold application in the hit identification process in early drug discovery.