 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCoupled Cluster con MōLe: Molecular Orbital Learning for Neural Wavefunctions
Feb 23, 2026Density functional theory (DFT) is the most widely used method for calculating molecular properties; however, its accuracy is often insufficient for quantitative predictions. Coupled-cluster (CC) theory is the most successful method for achieving accuracy beyond DFT and for predicting properties that closely align with experiment. It is known as the ''gold standard'' of quantum chemistry. Unfortunately, the high computational cost of CC limits its widespread applicability. In this work, we present the Molecular Orbital Learning (MōLe) architecture, an equivariant machine learning model that directly predicts CC's core mathematical objects, the excitation amplitudes, from the mean-field Hartree-Fock molecular orbitals as inputs. We test various aspects of our model and demonstrate its remarkable data efficiency and out-of-distribution generalization to larger molecules and off-equilibrium geometries, despite being trained only on small equilibrium geometries. Finally, we also examine its ability to reduce the number of cycles required to converge CC calculations. MōLe can set the foundations for high-accuracy wavefunction-based ML architectures to accelerate molecular design and complement force-field approaches.
El Agente Estructural: An Artificially Intelligent Molecular Editor
Feb 04, 2026We present El Agente Estructural, a multimodal, natural-language-driven geometry-generation and manipulation agent for autonomous chemistry and molecular modelling. Unlike molecular generation or editing via generative models, Estructural mimics how human experts directly manipulate molecular systems in three dimensions by integrating a comprehensive set of domain-informed tools and vision-language models. This design enables precise control over atomic or functional group replacements, atomic connectivity, and stereochemistry without the need to rebuild extensive core molecular frameworks. Through a series of representative case studies, we demonstrate that Estructural enables chemically meaningful geometry manipulation across a wide range of real-world scenarios. These include site-selective functionalization, ligand binding, ligand exchange, stereochemically controlled structure construction, isomer interconversion, fragment-level structural analysis, image-guided generation of structures from schematic reaction mechanisms, and mechanism-driven geometry generation and modification. These examples illustrate how multimodal reasoning, when combined with specialized geometry-aware tools, supports interactive and context-aware molecular modelling beyond structure generation. Looking forward, the integration of Estructural into El Agente Quntur, an autonomous multi-agent quantum chemistry platform, enhances its capabilities by adding sophisticated tools for the generation and editing of three-dimensional structures.
El Agente Quntur: A research collaborator agent for quantum chemistry
Feb 04, 2026Quantum chemistry is a foundational enabling tool for the fields of chemistry, materials science, computational biology and others. Despite of its power, the practical application of quantum chemistry simulations remains in the hands of qualified experts due to methodological complexity, software heterogeneity, and the need for informed interpretation of results. To bridge the accessibility gap for these tools and expand their reach to chemists with broader backgrounds, we introduce El Agente Quntur, a hierarchical, multi-agent AI system designed to operate not merely as an automation tool but as a research collaborator for computational quantum chemistry. Quntur was designed following three main strategies: i) elimination of hard-coded procedural policies in favour of reasoning-driven decisions, ii) construction of general and composable actions that facilitate generalization and efficiency, and iii) implementation of guided deep research to integrate abstract quantum-chemical reasoning across subdisciplines and a detailed understanding of the software's internal logic and syntax. Although instantiated in ORCA, these design principles are applicable to research agents more generally and easily expandable to additional quantum chemistry packages and beyond. Quntur supports the full range of calculations available in ORCA 6.0 and reasons over software documentation and scientific literature to plan, execute, adapt, and analyze in silico chemistry experiments following best practices. We discuss the advances and current bottlenecks in agentic systems operating at the research level in computational chemistry, and outline a roadmap toward a fully autonomous end-to-end computational chemistry research agent.
Quantum Computing-Enhanced Algorithm Unveils Novel Inhibitors for KRAS
Feb 13, 2024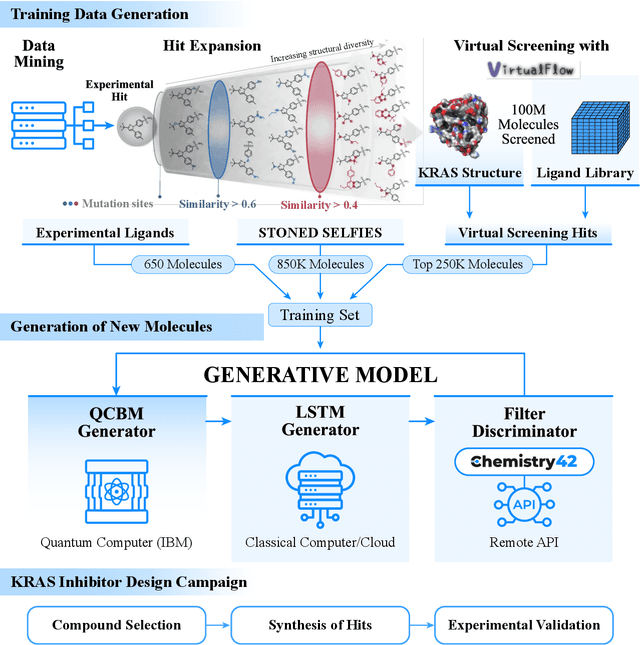
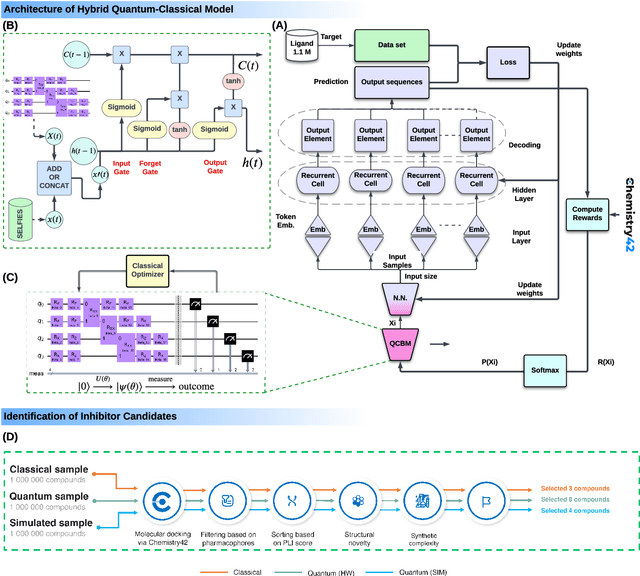
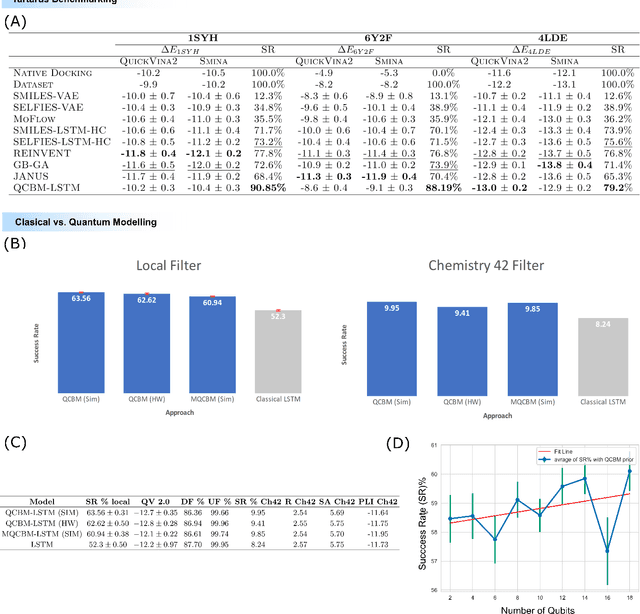
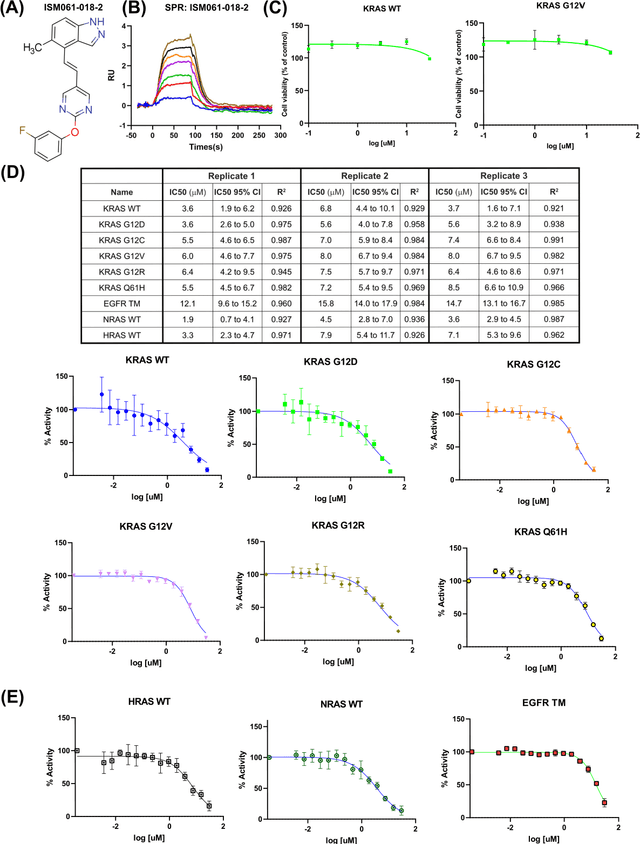
The discovery of small molecules with therapeutic potential is a long-standing challenge in chemistry and biology. Researchers have increasingly leveraged novel computational techniques to streamline the drug development process to increase hit rates and reduce the costs associated with bringing a drug to market. To this end, we introduce a quantum-classical generative model that seamlessly integrates the computational power of quantum algorithms trained on a 16-qubit IBM quantum computer with the established reliability of classical methods for designing small molecules. Our hybrid generative model was applied to designing new KRAS inhibitors, a crucial target in cancer therapy. We synthesized 15 promising molecules during our investigation and subjected them to experimental testing to assess their ability to engage with the target. Notably, among these candidates, two molecules, ISM061-018-2 and ISM061-22, each featuring unique scaffolds, stood out by demonstrating effective engagement with KRAS. ISM061-018-2 was identified as a broad-spectrum KRAS inhibitor, exhibiting a binding affinity to KRAS-G12D at $1.4 \mu M$. Concurrently, ISM061-22 exhibited specific mutant selectivity, displaying heightened activity against KRAS G12R and Q61H mutants. To our knowledge, this work shows for the first time the use of a quantum-generative model to yield experimentally confirmed biological hits, showcasing the practical potential of quantum-assisted drug discovery to produce viable therapeutics. Moreover, our findings reveal that the efficacy of distribution learning correlates with the number of qubits utilized, underlining the scalability potential of quantum computing resources. Overall, we anticipate our results to be a stepping stone towards developing more advanced quantum generative models in drug discovery.