 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeQuantum Computing-Enhanced Algorithm Unveils Novel Inhibitors for KRAS
Feb 13, 2024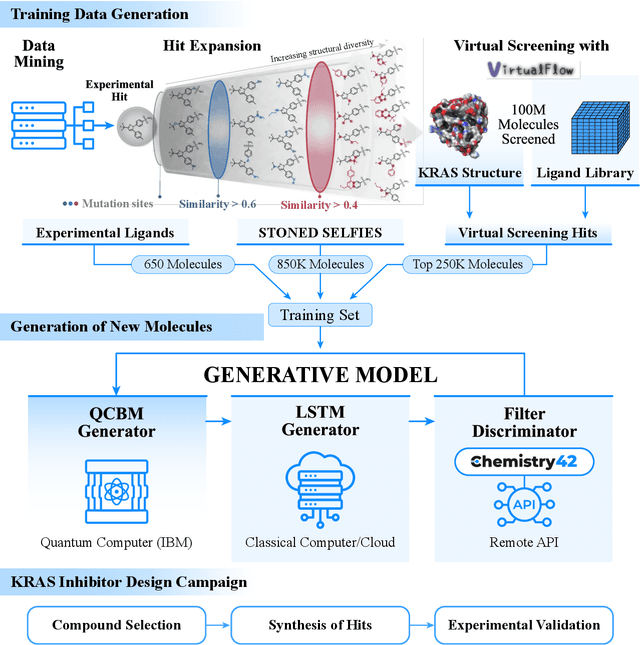
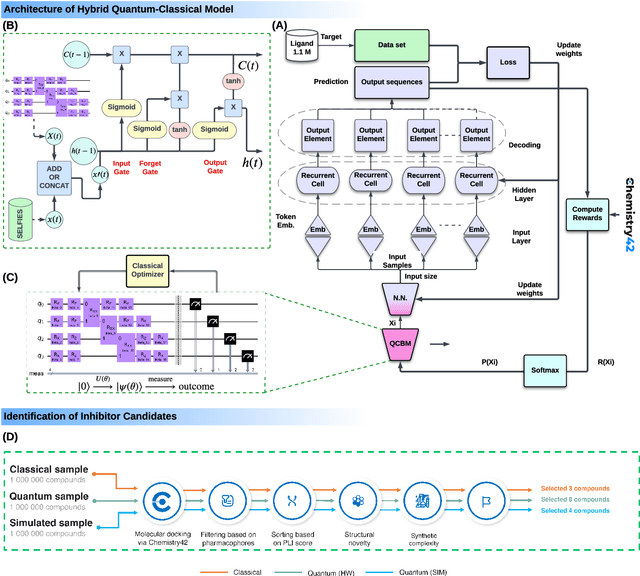
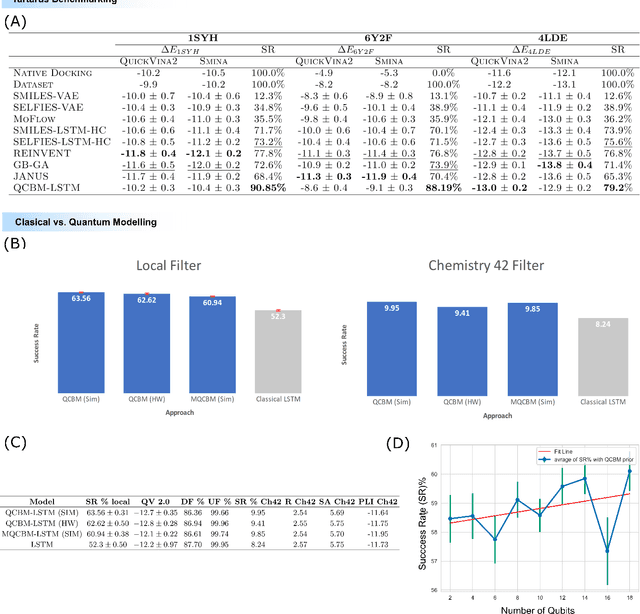
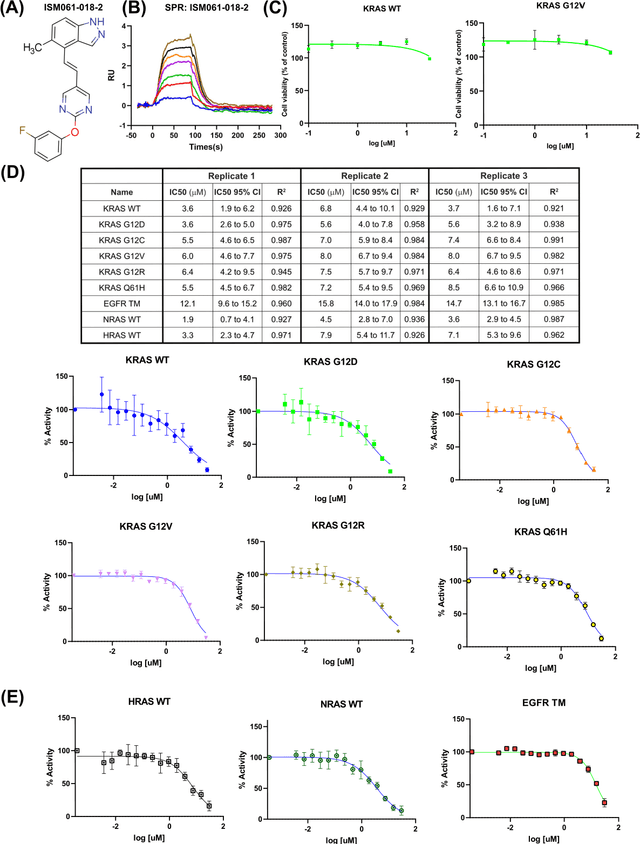
The discovery of small molecules with therapeutic potential is a long-standing challenge in chemistry and biology. Researchers have increasingly leveraged novel computational techniques to streamline the drug development process to increase hit rates and reduce the costs associated with bringing a drug to market. To this end, we introduce a quantum-classical generative model that seamlessly integrates the computational power of quantum algorithms trained on a 16-qubit IBM quantum computer with the established reliability of classical methods for designing small molecules. Our hybrid generative model was applied to designing new KRAS inhibitors, a crucial target in cancer therapy. We synthesized 15 promising molecules during our investigation and subjected them to experimental testing to assess their ability to engage with the target. Notably, among these candidates, two molecules, ISM061-018-2 and ISM061-22, each featuring unique scaffolds, stood out by demonstrating effective engagement with KRAS. ISM061-018-2 was identified as a broad-spectrum KRAS inhibitor, exhibiting a binding affinity to KRAS-G12D at $1.4 \mu M$. Concurrently, ISM061-22 exhibited specific mutant selectivity, displaying heightened activity against KRAS G12R and Q61H mutants. To our knowledge, this work shows for the first time the use of a quantum-generative model to yield experimentally confirmed biological hits, showcasing the practical potential of quantum-assisted drug discovery to produce viable therapeutics. Moreover, our findings reveal that the efficacy of distribution learning correlates with the number of qubits utilized, underlining the scalability potential of quantum computing resources. Overall, we anticipate our results to be a stepping stone towards developing more advanced quantum generative models in drug discovery.
Recent Developments in Structure-Based Virtual Screening Approaches
Nov 06, 2022

Drug development is a wide scientific field that faces many challenges these days. Among them are extremely high development costs, long development times, as well as a low number of new drugs that are approved each year. To solve these problems, new and innovate technologies are needed that make the drug discovery process of small-molecules more time and cost-efficient, and which allow to target previously undruggable target classes such as protein-protein interactions. Structure-based virtual screenings have become a leading contender in this context. In this review, we give an introduction to the foundations of structure-based virtual screenings, and survey their progress in the past few years. We outline key principles, recent success stories, new methods, available software, and promising future research directions. Virtual screenings have an enormous potential for the development of new small-molecule drugs, and are already starting to transform early-stage drug discovery.