 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeQuantum Computing-Enhanced Algorithm Unveils Novel Inhibitors for KRAS
Feb 13, 2024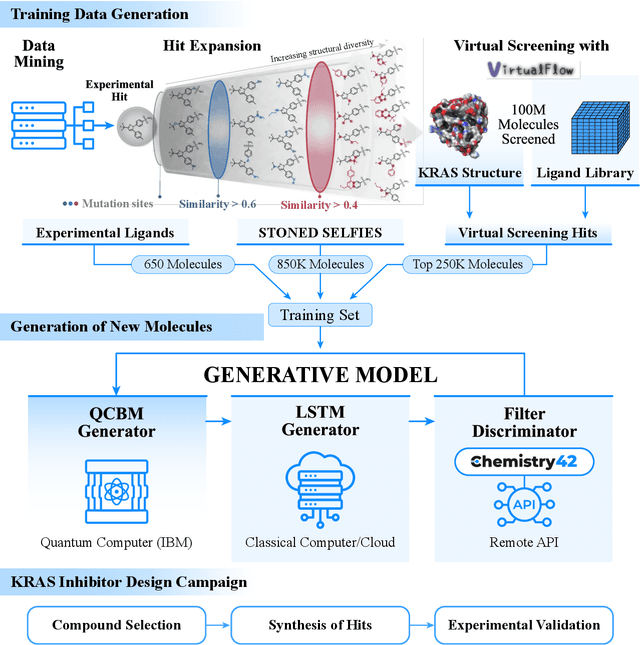
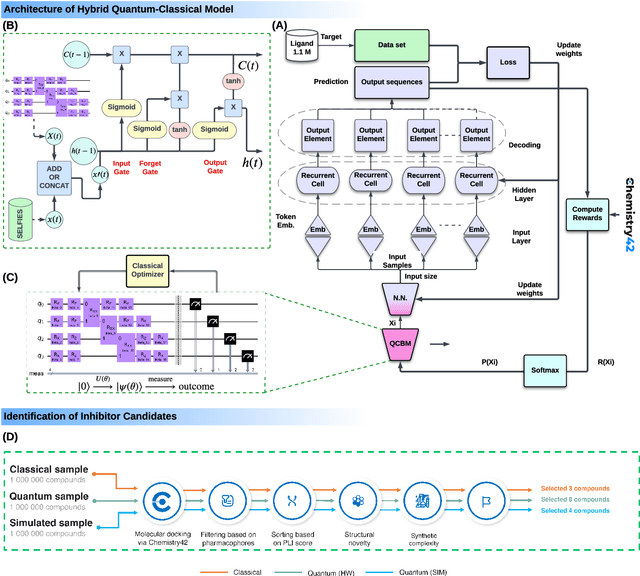
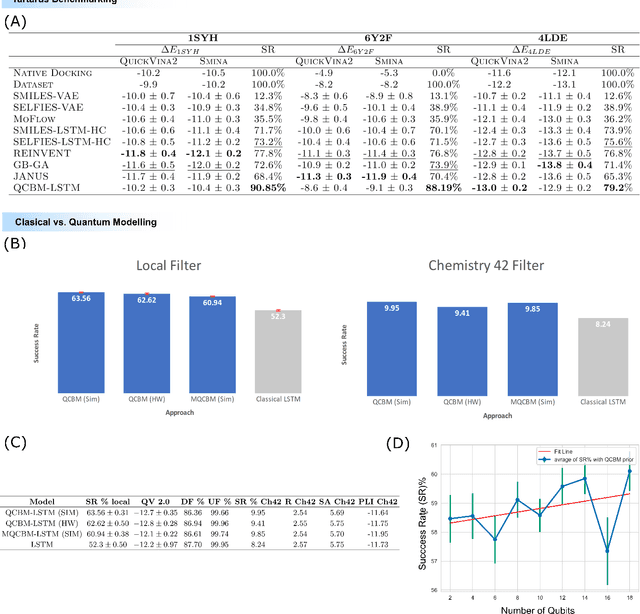
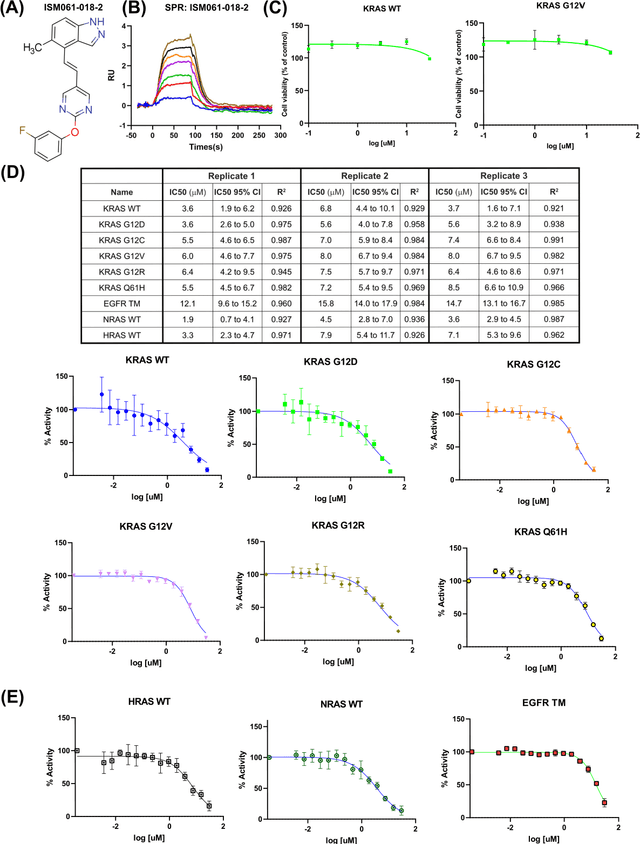
The discovery of small molecules with therapeutic potential is a long-standing challenge in chemistry and biology. Researchers have increasingly leveraged novel computational techniques to streamline the drug development process to increase hit rates and reduce the costs associated with bringing a drug to market. To this end, we introduce a quantum-classical generative model that seamlessly integrates the computational power of quantum algorithms trained on a 16-qubit IBM quantum computer with the established reliability of classical methods for designing small molecules. Our hybrid generative model was applied to designing new KRAS inhibitors, a crucial target in cancer therapy. We synthesized 15 promising molecules during our investigation and subjected them to experimental testing to assess their ability to engage with the target. Notably, among these candidates, two molecules, ISM061-018-2 and ISM061-22, each featuring unique scaffolds, stood out by demonstrating effective engagement with KRAS. ISM061-018-2 was identified as a broad-spectrum KRAS inhibitor, exhibiting a binding affinity to KRAS-G12D at $1.4 \mu M$. Concurrently, ISM061-22 exhibited specific mutant selectivity, displaying heightened activity against KRAS G12R and Q61H mutants. To our knowledge, this work shows for the first time the use of a quantum-generative model to yield experimentally confirmed biological hits, showcasing the practical potential of quantum-assisted drug discovery to produce viable therapeutics. Moreover, our findings reveal that the efficacy of distribution learning correlates with the number of qubits utilized, underlining the scalability potential of quantum computing resources. Overall, we anticipate our results to be a stepping stone towards developing more advanced quantum generative models in drug discovery.
Recent advances in the Self-Referencing Embedding Strings (SELFIES) library
Feb 07, 2023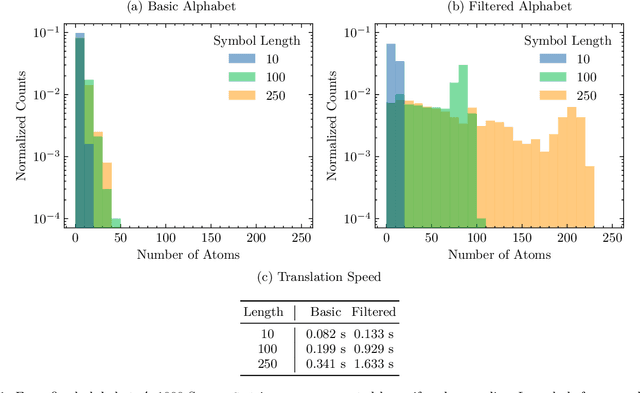
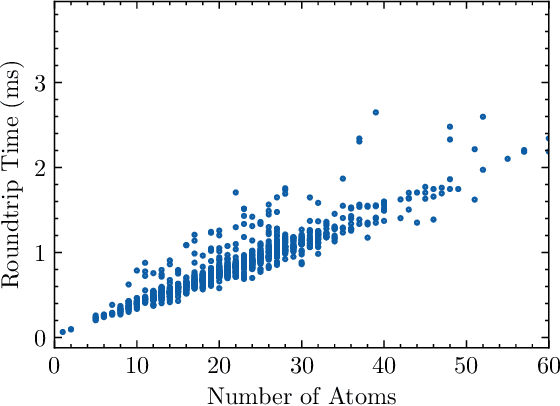

String-based molecular representations play a crucial role in cheminformatics applications, and with the growing success of deep learning in chemistry, have been readily adopted into machine learning pipelines. However, traditional string-based representations such as SMILES are often prone to syntactic and semantic errors when produced by generative models. To address these problems, a novel representation, SELF-referencIng Embedded Strings (SELFIES), was proposed that is inherently 100% robust, alongside an accompanying open-source implementation. Since then, we have generalized SELFIES to support a wider range of molecules and semantic constraints and streamlined its underlying grammar. We have implemented this updated representation in subsequent versions of \selfieslib, where we have also made major advances with respect to design, efficiency, and supported features. Hence, we present the current status of \selfieslib (version 2.1.1) in this manuscript.
On scientific understanding with artificial intelligence
Apr 04, 2022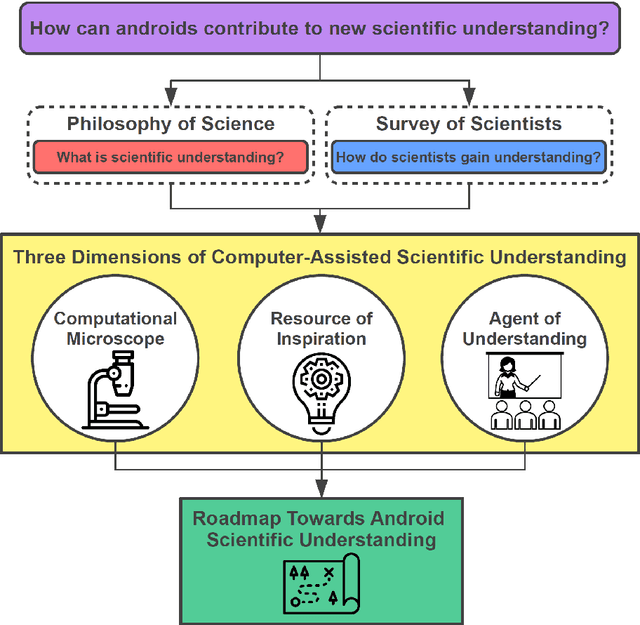
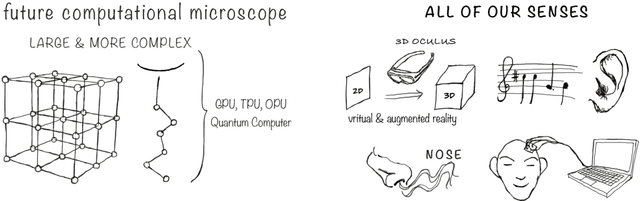
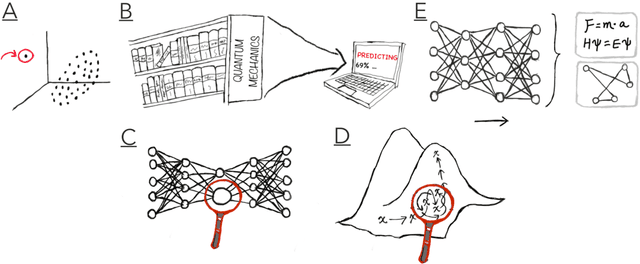
Imagine an oracle that correctly predicts the outcome of every particle physics experiment, the products of every chemical reaction, or the function of every protein. Such an oracle would revolutionize science and technology as we know them. However, as scientists, we would not be satisfied with the oracle itself. We want more. We want to comprehend how the oracle conceived these predictions. This feat, denoted as scientific understanding, has frequently been recognized as the essential aim of science. Now, the ever-growing power of computers and artificial intelligence poses one ultimate question: How can advanced artificial systems contribute to scientific understanding or achieve it autonomously? We are convinced that this is not a mere technical question but lies at the core of science. Therefore, here we set out to answer where we are and where we can go from here. We first seek advice from the philosophy of science to understand scientific understanding. Then we review the current state of the art, both from literature and by collecting dozens of anecdotes from scientists about how they acquired new conceptual understanding with the help of computers. Those combined insights help us to define three dimensions of android-assisted scientific understanding: The android as a I) computational microscope, II) resource of inspiration and the ultimate, not yet existent III) agent of understanding. For each dimension, we explain new avenues to push beyond the status quo and unleash the full power of artificial intelligence's contribution to the central aim of science. We hope our perspective inspires and focuses research towards androids that get new scientific understanding and ultimately bring us closer to true artificial scientists.
SELFIES and the future of molecular string representations
Mar 31, 2022
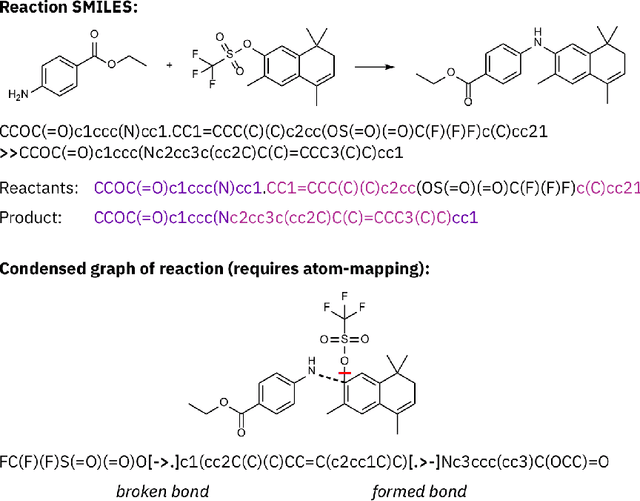
Artificial intelligence (AI) and machine learning (ML) are expanding in popularity for broad applications to challenging tasks in chemistry and materials science. Examples include the prediction of properties, the discovery of new reaction pathways, or the design of new molecules. The machine needs to read and write fluently in a chemical language for each of these tasks. Strings are a common tool to represent molecular graphs, and the most popular molecular string representation, SMILES, has powered cheminformatics since the late 1980s. However, in the context of AI and ML in chemistry, SMILES has several shortcomings -- most pertinently, most combinations of symbols lead to invalid results with no valid chemical interpretation. To overcome this issue, a new language for molecules was introduced in 2020 that guarantees 100\% robustness: SELFIES (SELF-referencIng Embedded Strings). SELFIES has since simplified and enabled numerous new applications in chemistry. In this manuscript, we look to the future and discuss molecular string representations, along with their respective opportunities and challenges. We propose 16 concrete Future Projects for robust molecular representations. These involve the extension toward new chemical domains, exciting questions at the interface of AI and robust languages and interpretability for both humans and machines. We hope that these proposals will inspire several follow-up works exploiting the full potential of molecular string representations for the future of AI in chemistry and materials science.
JANUS: Parallel Tempered Genetic Algorithm Guided by Deep Neural Networks for Inverse Molecular Design
Jun 07, 2021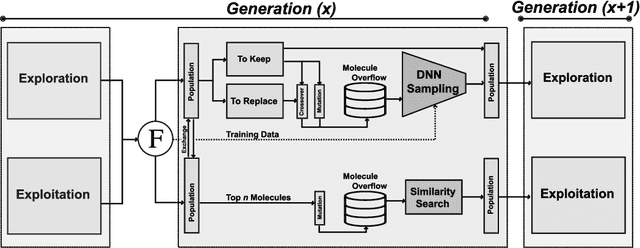

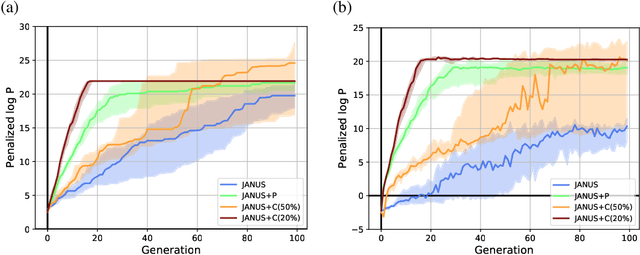
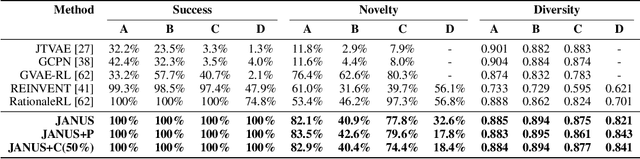
Inverse molecular design, i.e., designing molecules with specific target properties, can be posed as an optimization problem. High-dimensional optimization tasks in the natural sciences are commonly tackled via population-based metaheuristic optimization algorithms such as evolutionary algorithms. However, expensive property evaluation, which is often required, can limit the widespread use of such approaches as the associated cost can become prohibitive. Herein, we present JANUS, a genetic algorithm that is inspired by parallel tempering. It propagates two populations, one for exploration and another for exploitation, improving optimization by reducing expensive property evaluations. Additionally, JANUS is augmented by a deep neural network that approximates molecular properties via active learning for enhanced sampling of the chemical space. Our method uses the SELFIES molecular representation and the STONED algorithm for the efficient generation of structures, and outperforms other generative models in common inverse molecular design tasks achieving state-of-the-art performance.
Assigning Confidence to Molecular Property Prediction
Feb 23, 2021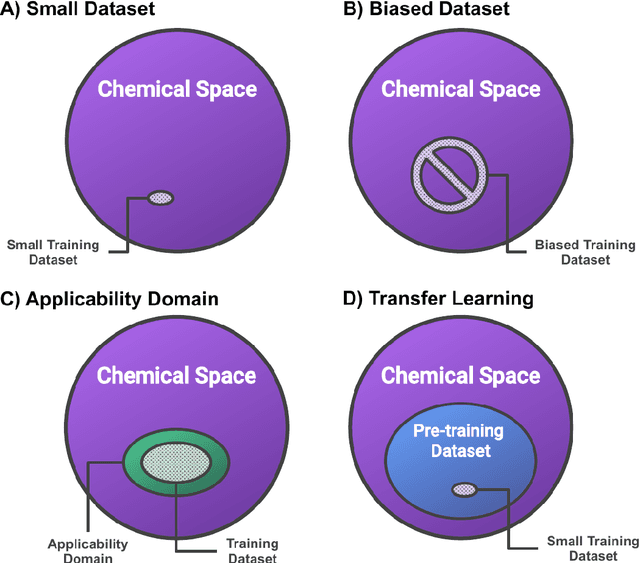
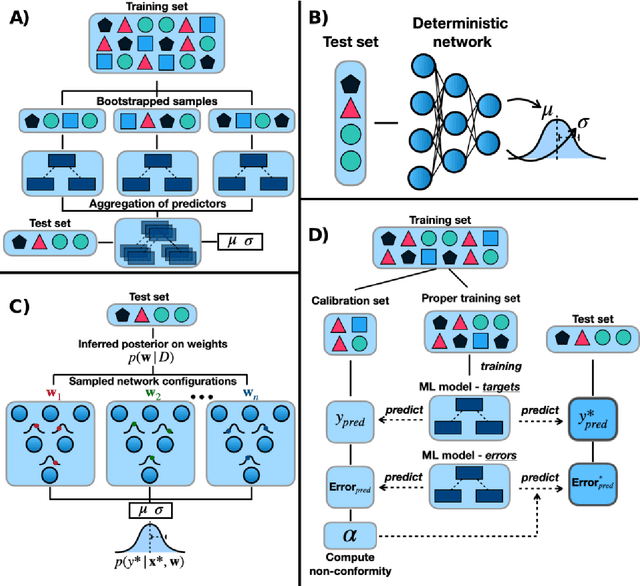
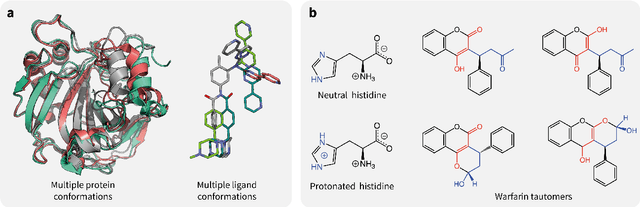
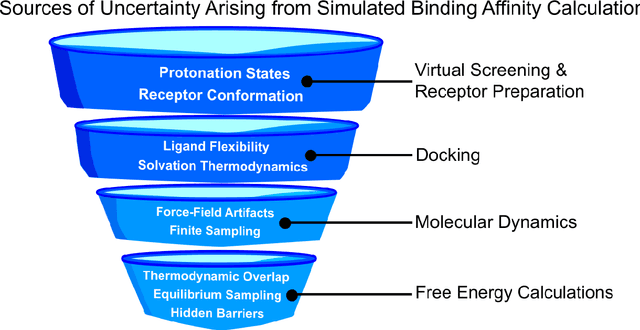
Introduction: Computational modeling has rapidly advanced over the last decades, especially to predict molecular properties for chemistry, material science and drug design. Recently, machine learning techniques have emerged as a powerful and cost-effective strategy to learn from existing datasets and perform predictions on unseen molecules. Accordingly, the explosive rise of data-driven techniques raises an important question: What confidence can be assigned to molecular property predictions and what techniques can be used for that purpose? Areas covered: In this work, we discuss popular strategies for predicting molecular properties relevant to drug design, their corresponding uncertainty sources and methods to quantify uncertainty and confidence. First, our considerations for assessing confidence begin with dataset bias and size, data-driven property prediction and feature design. Next, we discuss property simulation via molecular docking, and free-energy simulations of binding affinity in detail. Lastly, we investigate how these uncertainties propagate to generative models, as they are usually coupled with property predictors. Expert opinion: Computational techniques are paramount to reduce the prohibitive cost and timing of brute-force experimentation when exploring the enormous chemical space. We believe that assessing uncertainty in property prediction models is essential whenever closed-loop drug design campaigns relying on high-throughput virtual screening are deployed. Accordingly, considering sources of uncertainty leads to better-informed experimental validations, more reliable predictions and to more realistic expectations of the entire workflow. Overall, this increases confidence in the predictions and designs and, ultimately, accelerates drug design.
Curiosity in exploring chemical space: Intrinsic rewards for deep molecular reinforcement learning
Dec 17, 2020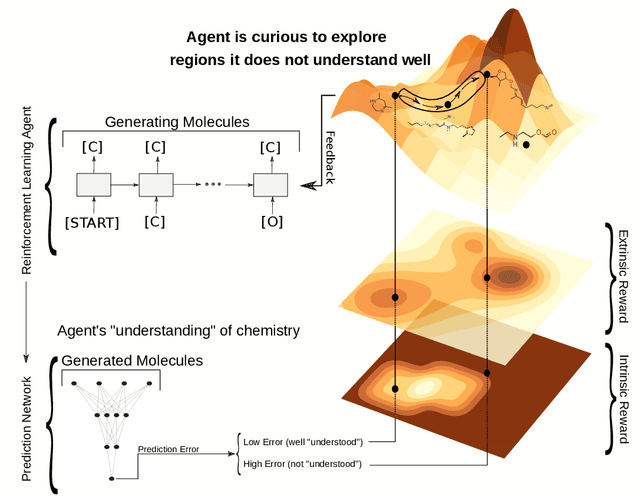
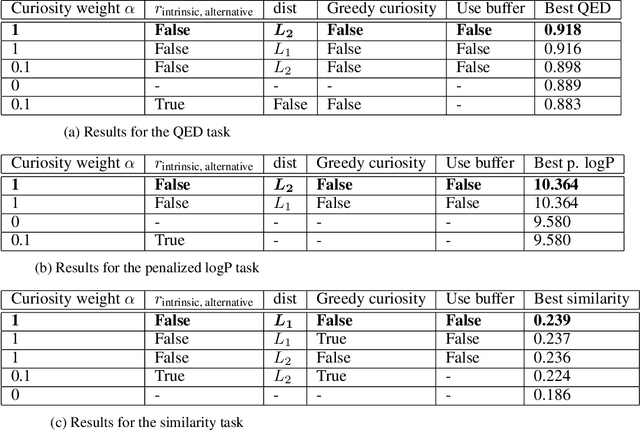
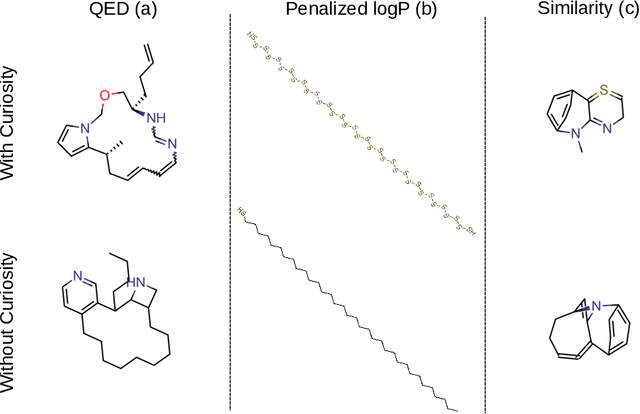
Computer-aided design of molecules has the potential to disrupt the field of drug and material discovery. Machine learning, and deep learning, in particular, have been topics where the field has been developing at a rapid pace. Reinforcement learning is a particularly promising approach since it allows for molecular design without prior knowledge. However, the search space is vast and efficient exploration is desirable when using reinforcement learning agents. In this study, we propose an algorithm to aid efficient exploration. The algorithm is inspired by a concept known in the literature as curiosity. We show on three benchmarks that a curious agent finds better performing molecules. This indicates an exciting new research direction for reinforcement learning agents that can explore the chemical space out of their own motivation. This has the potential to eventually lead to unexpected new molecules that no human has thought about so far.
Augmenting Genetic Algorithms with Deep Neural Networks for Exploring the Chemical Space
Sep 30, 2019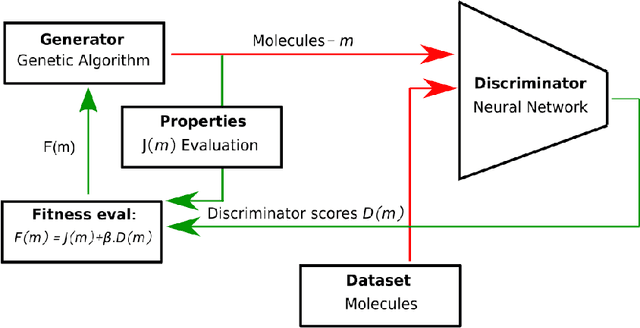
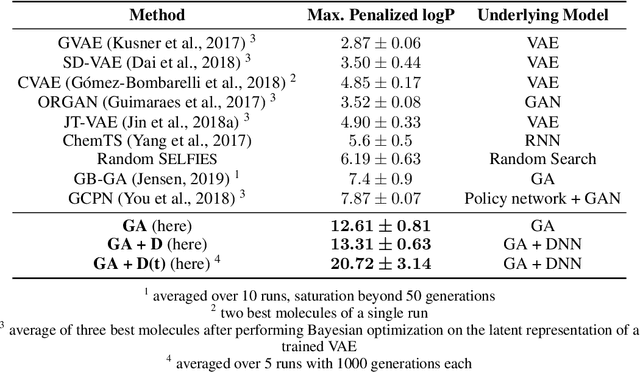
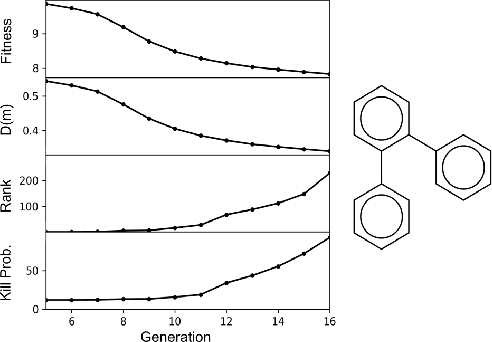
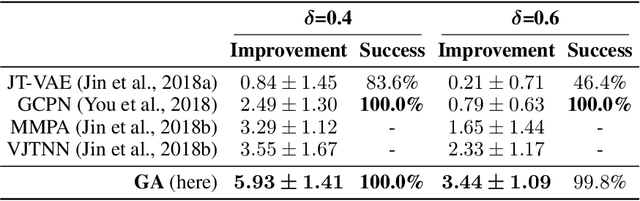
Challenges in natural sciences can often be phrased as optimization problems. Machine learning techniques have recently been applied to solve such problems. One example in chemistry is the design of tailor-made organic materials and molecules, which requires efficient methods to explore the chemical space. We present a genetic algorithm (GA) that is enhanced with a neural network (DNN) based discriminator model to improve the diversity of generated molecules and at the same time steer the GA. We show that our algorithm outperforms other generative models in optimization tasks. We furthermore present a way to increase interpretability of genetic algorithms, which helped us to derive design principles.
SELFIES: a robust representation of semantically constrained graphs with an example application in chemistry
May 31, 2019
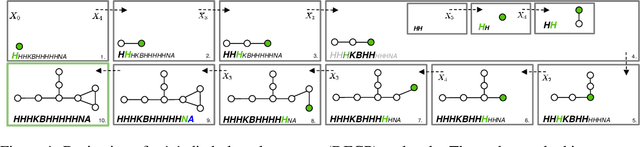
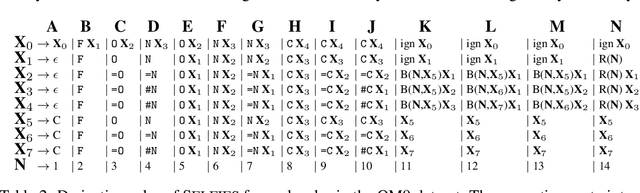
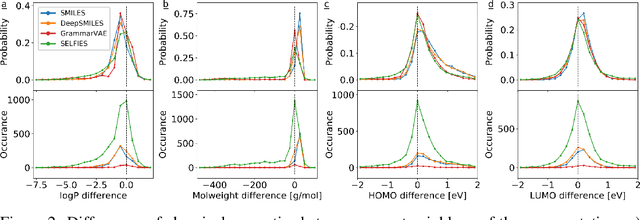
Graphs are ideal representations of complex, relational information. Their applications span diverse areas of science and engineering, such as Feynman diagrams in fundamental physics, the structures of molecules in chemistry or transport systems in urban planning. Recently, many of these examples turned into the spotlight as applications of machine learning (ML). There, common challenges to the successful deployment of ML are domain-specific constraints, which lead to semantically constrained graphs. While much progress has been achieved in the generation of valid graphs for domain- and model-specific applications, a general approach has not been demonstrated yet. Here, we present a general-purpose, sequence-based, robust representation of semantically constrained graphs, which we call SELFIES (SELF-referencIng Embedded Strings). SELFIES are based on a Chomsky type-2 grammar, augmented with two self-referencing functions. We demonstrate their applicability to represent chemical compound structures and compare them to perhaps the most popular 2D representation, SMILES, and other important baselines. We find stronger robustness against character mutations while still maintaining similar chemical properties. Even entirely random SELFIES produce semantically valid graphs in most of the cases. As feature representation in variational autoencoders, SELFIES provide a substantial improvement in the task of in reconstruction, validity, and diversity. We anticipate that SELFIES allow for direct applications in ML, without the need for domain-specific adaptation of model architectures. SELFIES are not limited to the structures of small molecules, and we show how to apply them to two other examples from the sciences: representations of DNA and interaction graphs for quantum mechanical experiments.