 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeChemBERTa-2: Towards Chemical Foundation Models
Sep 05, 2022
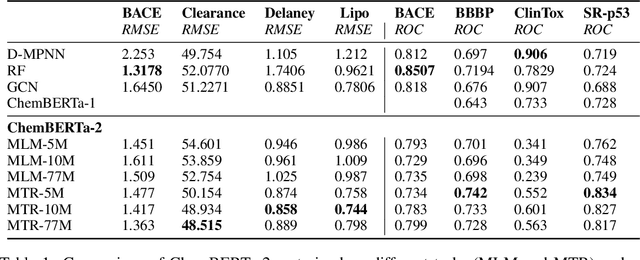
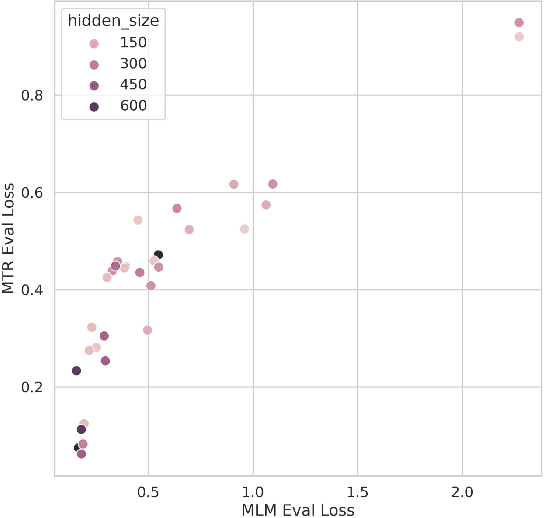
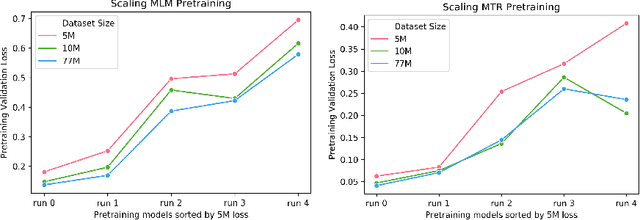
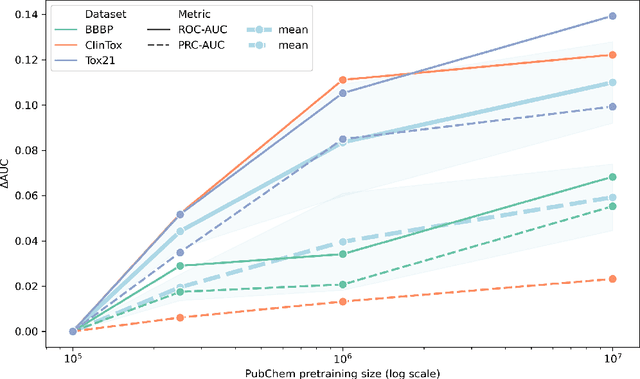
Large pretrained models such as GPT-3 have had tremendous impact on modern natural language processing by leveraging self-supervised learning to learn salient representations that can be used to readily finetune on a wide variety of downstream tasks. We investigate the possibility of transferring such advances to molecular machine learning by building a chemical foundation model, ChemBERTa-2, using the language of SMILES. While labeled data for molecular prediction tasks is typically scarce, libraries of SMILES strings are readily available. In this work, we build upon ChemBERTa by optimizing the pretraining process. We compare multi-task and self-supervised pretraining by varying hyperparameters and pretraining dataset size, up to 77M compounds from PubChem. To our knowledge, the 77M set constitutes one of the largest datasets used for molecular pretraining to date. We find that with these pretraining improvements, we are competitive with existing state-of-the-art architectures on the MoleculeNet benchmark suite. We analyze the degree to which improvements in pretraining translate to improvement on downstream tasks.
Assigning Confidence to Molecular Property Prediction
Feb 23, 2021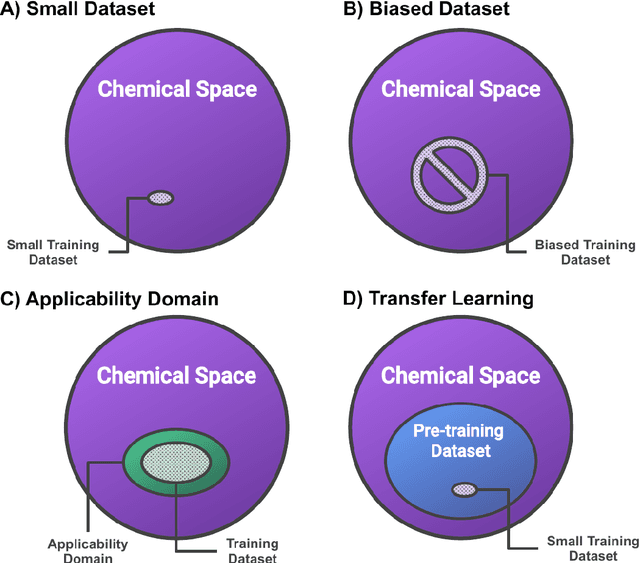
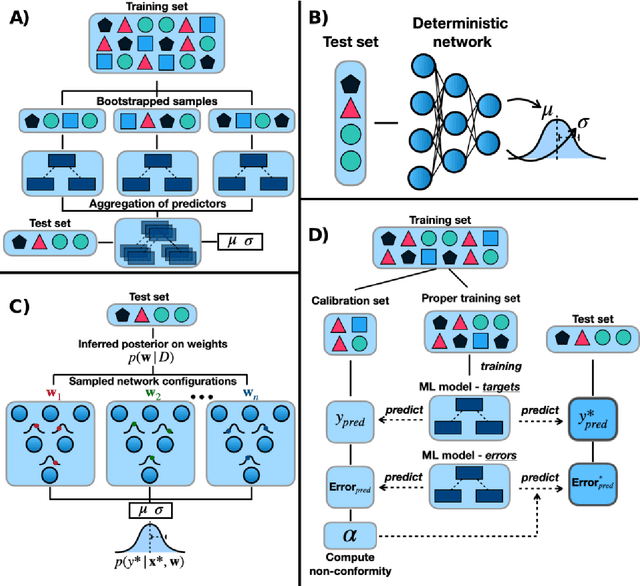
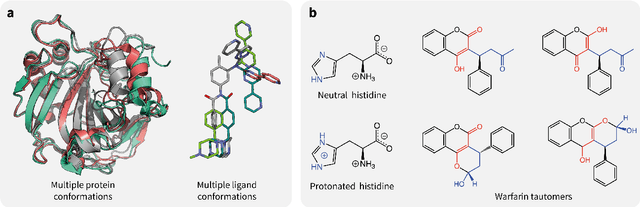
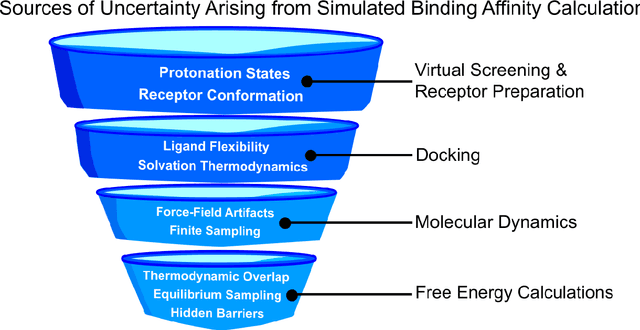
Introduction: Computational modeling has rapidly advanced over the last decades, especially to predict molecular properties for chemistry, material science and drug design. Recently, machine learning techniques have emerged as a powerful and cost-effective strategy to learn from existing datasets and perform predictions on unseen molecules. Accordingly, the explosive rise of data-driven techniques raises an important question: What confidence can be assigned to molecular property predictions and what techniques can be used for that purpose? Areas covered: In this work, we discuss popular strategies for predicting molecular properties relevant to drug design, their corresponding uncertainty sources and methods to quantify uncertainty and confidence. First, our considerations for assessing confidence begin with dataset bias and size, data-driven property prediction and feature design. Next, we discuss property simulation via molecular docking, and free-energy simulations of binding affinity in detail. Lastly, we investigate how these uncertainties propagate to generative models, as they are usually coupled with property predictors. Expert opinion: Computational techniques are paramount to reduce the prohibitive cost and timing of brute-force experimentation when exploring the enormous chemical space. We believe that assessing uncertainty in property prediction models is essential whenever closed-loop drug design campaigns relying on high-throughput virtual screening are deployed. Accordingly, considering sources of uncertainty leads to better-informed experimental validations, more reliable predictions and to more realistic expectations of the entire workflow. Overall, this increases confidence in the predictions and designs and, ultimately, accelerates drug design.
ChemBERTa: Large-Scale Self-Supervised Pretraining for Molecular Property Prediction
Oct 23, 2020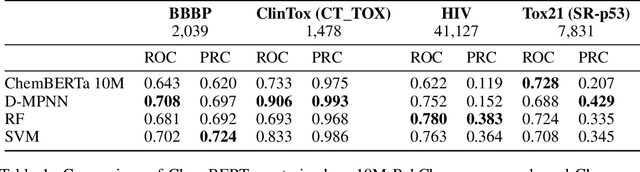
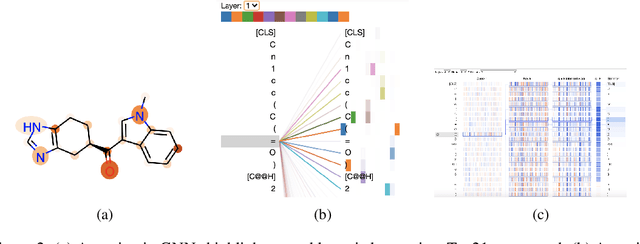
GNNs and chemical fingerprints are the predominant approaches to representing molecules for property prediction. However, in NLP, transformers have become the de-facto standard for representation learning thanks to their strong downstream task transfer. In parallel, the software ecosystem around transformers is maturing rapidly, with libraries like HuggingFace and BertViz enabling streamlined training and introspection. In this work, we make one of the first attempts to systematically evaluate transformers on molecular property prediction tasks via our ChemBERTa model. ChemBERTa scales well with pretraining dataset size, offering competitive downstream performance on MoleculeNet and useful attention-based visualization modalities. Our results suggest that transformers offer a promising avenue of future work for molecular representation learning and property prediction. To facilitate these efforts, we release a curated dataset of 77M SMILES from PubChem suitable for large-scale self-supervised pretraining.