 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA fully differentiable framework for training proxy Exchange Correlation Functionals for periodic systems
Feb 17, 2026Density Functional Theory (DFT) is widely used for first-principles simulations in chemistry and materials science, but its computational cost remains a key limitation for large systems. Motivated by recent advances in ML-based exchange-correlation (XC) functionals, this paper introduces a differentiable framework that integrates machine learning models into density functional theory (DFT) for solids and other periodic systems. The framework defines a clean API for neural network models that can act as drop in replacements for conventional exchange-correlation (XC) functionals and enables gradients to flow through the full self-consistent DFT workflow. The framework is implemented in Python using a PyTorch backend, making it fully differentiable and easy to use with standard deep learning tools. We integrate the implementation with the DeepChem library to promote the reuse of established models and to lower the barrier for experimentation. In initial benchmarks against established electronic structure packages (GPAW and PySCF), our models achieve relative errors on the order of 5-10%.
Protect$^*$: Steerable Retrosynthesis through Neuro-Symbolic State Encoding
Feb 13, 2026Large Language Models (LLMs) have shown remarkable potential in scientific domains like retrosynthesis; yet, they often lack the fine-grained control necessary to navigate complex problem spaces without error. A critical challenge is directing an LLM to avoid specific, chemically sensitive sites on a molecule - a task where unconstrained generation can lead to invalid or undesirable synthetic pathways. In this work, we introduce Protect$^*$, a neuro-symbolic framework that grounds the generative capabilities of Large Language Models (LLMs) in rigorous chemical logic. Our approach combines automated rule-based reasoning - using a comprehensive database of 55+ SMARTS patterns and 40+ characterized protecting groups - with the generative intuition of neural models. The system operates via a hybrid architecture: an ``automatic mode'' where symbolic logic deterministically identifies and guards reactive sites, and a ``human-in-the-loop mode'' that integrates expert strategic constraints. Through ``active state tracking,'' we inject hard symbolic constraints into the neural inference process via a dedicated protection state linked to canonical atom maps. We demonstrate this neuro-symbolic approach through case studies on complex natural products, including the discovery of a novel synthetic pathway for Erythromycin B, showing that grounding neural generation in symbolic logic enables reliable, expert-level autonomy.
Open-Source Protein Language Models for Function Prediction and Protein Design
Dec 18, 2024Protein language models (PLMs) have shown promise in improving the understanding of protein sequences, contributing to advances in areas such as function prediction and protein engineering. However, training these models from scratch requires significant computational resources, limiting their accessibility. To address this, we integrate a PLM into DeepChem, an open-source framework for computational biology and chemistry, to provide a more accessible platform for protein-related tasks. We evaluate the performance of the integrated model on various protein prediction tasks, showing that it achieves reasonable results across benchmarks. Additionally, we present an exploration of generating plastic-degrading enzyme candidates using the model's embeddings and latent space manipulation techniques. While the results suggest that further refinement is needed, this approach provides a foundation for future work in enzyme design. This study aims to facilitate the use of PLMs in research fields like synthetic biology and environmental sustainability, even for those with limited computational resources.
Open source Differentiable ODE Solving Infrastructure
Nov 29, 2024Ordinary Differential Equations (ODEs) are widely used in physics, chemistry, and biology to model dynamic systems, including reaction kinetics, population dynamics, and biological processes. In this work, we integrate GPU-accelerated ODE solvers into the open-source DeepChem framework, making these tools easily accessible. These solvers support multiple numerical methods and are fully differentiable, enabling easy integration into more complex differentiable programs. We demonstrate the capabilities of our implementation through experiments on Lotka-Volterra predator-prey dynamics, pharmacokinetic compartment models, neural ODEs, and solving PDEs using reaction-diffusion equations. Our solvers achieved high accuracy with mean squared errors ranging from $10^{-4}$ to $10^{-6}$ and showed scalability in solving large systems with up to 100 compartments.
A Modular Open Source Framework for Genomic Variant Calling
Nov 18, 2024Variant calling is a fundamental task in genomic research, essential for detecting genetic variations such as single nucleotide polymorphisms (SNPs) and insertions or deletions (indels). This paper presents an enhancement to DeepChem, a widely used open-source drug discovery framework, through the integration of DeepVariant. In particular, we introduce a variant calling pipeline that leverages DeepVariant's convolutional neural network (CNN) architecture to improve the accuracy and reliability of variant detection. The implemented pipeline includes stages for realignment of sequencing reads, candidate variant detection, and pileup image generation, followed by variant classification using a modified Inception v3 model. Our work adds a modular and extensible variant calling framework to the DeepChem framework and enables future work integrating DeepChem's drug discovery infrastructure more tightly with bioinformatics pipelines.
Open Source Infrastructure for Automatic Cell Segmentation
Sep 12, 2024Automated cell segmentation is crucial for various biological and medical applications, facilitating tasks like cell counting, morphology analysis, and drug discovery. However, manual segmentation is time-consuming and prone to subjectivity, necessitating robust automated methods. This paper presents open-source infrastructure, utilizing the UNet model, a deep-learning architecture noted for its effectiveness in image segmentation tasks. This implementation is integrated into the open-source DeepChem package, enhancing accessibility and usability for researchers and practitioners. The resulting tool offers a convenient and user-friendly interface, reducing the barrier to entry for cell segmentation while maintaining high accuracy. Additionally, we benchmark this model against various datasets, demonstrating its robustness and versatility across different imaging conditions and cell types.
Open-Source Molecular Processing Pipeline for Generating Molecules
Aug 12, 2024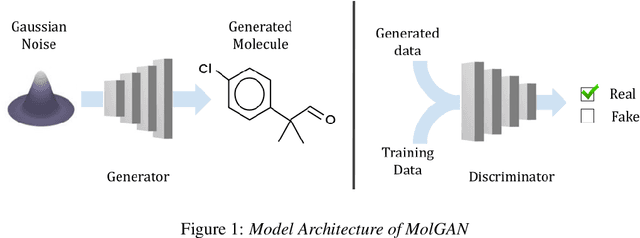
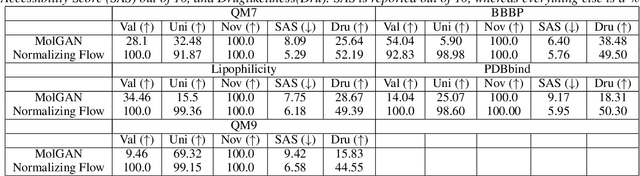
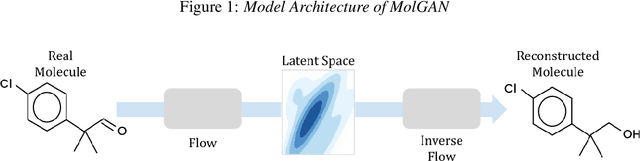
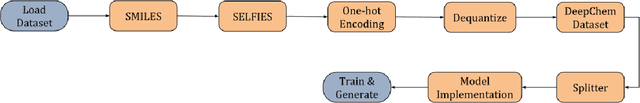
Generative models for molecules have shown considerable promise for use in computational chemistry, but remain difficult to use for non-experts. For this reason, we introduce open-source infrastructure for easily building generative molecular models into the widely used DeepChem [Ramsundar et al., 2019] library with the aim of creating a robust and reusable molecular generation pipeline. In particular, we add high quality PyTorch [Paszke et al., 2019] implementations of the Molecular Generative Adversarial Networks (MolGAN) [Cao and Kipf, 2022] and Normalizing Flows [Papamakarios et al., 2021]. Our implementations show strong performance comparable with past work [Kuznetsov and Polykovskiy, 2021, Cao and Kipf, 2022].
Self-supervised Pretraining for Partial Differential Equations
Jul 03, 2024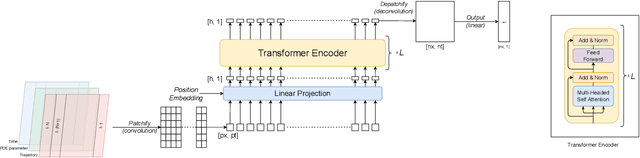
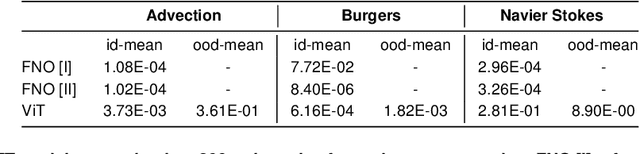
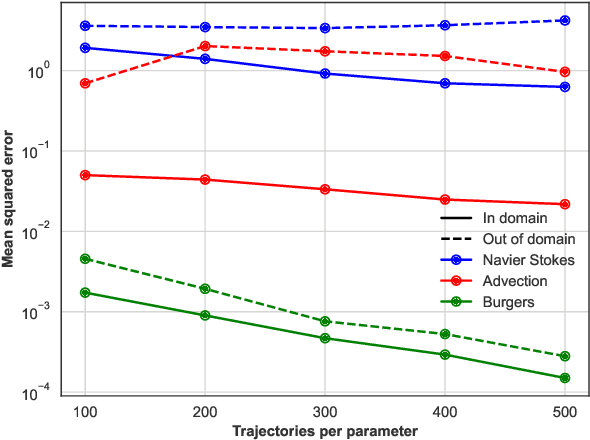

In this work, we describe a novel approach to building a neural PDE solver leveraging recent advances in transformer based neural network architectures. Our model can provide solutions for different values of PDE parameters without any need for retraining the network. The training is carried out in a self-supervised manner, similar to pretraining approaches applied in language and vision tasks. We hypothesize that the model is in effect learning a family of operators (for multiple parameters) mapping the initial condition to the solution of the PDE at any future time step t. We compare this approach with the Fourier Neural Operator (FNO), and demonstrate that it can generalize over the space of PDE parameters, despite having a higher prediction error for individual parameter values compared to the FNO. We show that performance on a specific parameter can be improved by finetuning the model with very small amounts of data. We also demonstrate that the model scales with data as well as model size.
Open-Source Fermionic Neural Networks with Ionic Charge Initialization
Jan 16, 2024Finding accurate solutions to the electronic Schr\"odinger equation plays an important role in discovering important molecular and material energies and characteristics. Consequently, solving systems with large numbers of electrons has become increasingly important. Variational Monte Carlo (VMC) methods, especially those approximated through deep neural networks, are promising in this regard. In this paper, we aim to integrate one such model called the FermiNet, a post-Hartree-Fock (HF) Deep Neural Network (DNN) model, into a standard and widely used open source library, DeepChem. We also propose novel initialization techniques to overcome the difficulties associated with the assignment of excess or lack of electrons for ions.
Differentiable Chemical Physics by Geometric Deep Learning for Gradient-based Property Optimization of Mixtures
Oct 03, 2023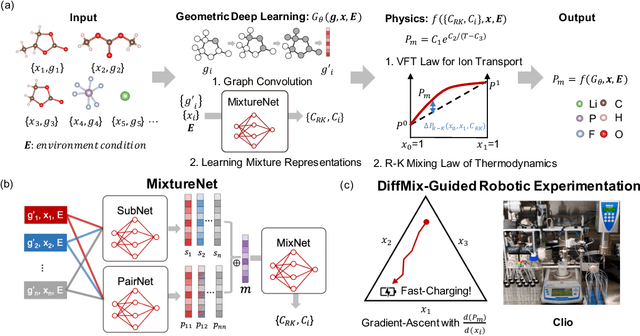
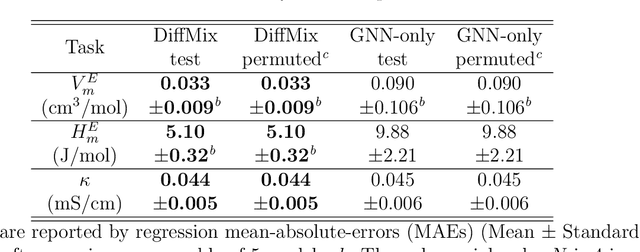
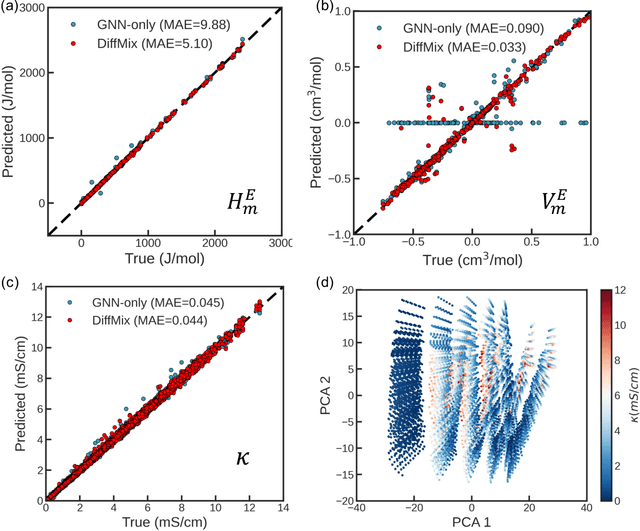
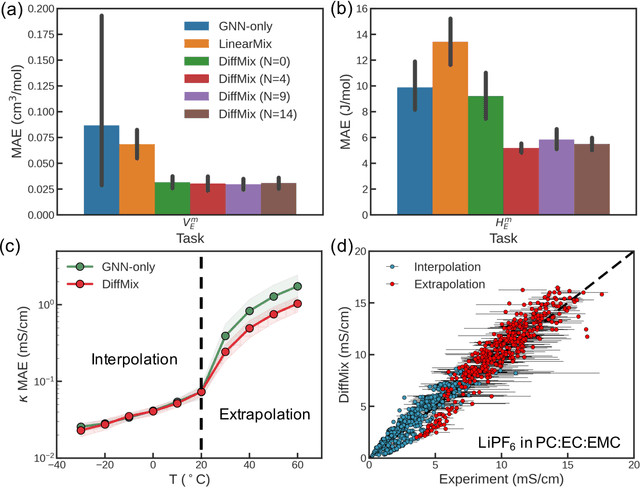
Chemical mixtures, satisfying multi-objective performance metrics and constraints, enable their use in chemical processes and electrochemical devices. In this work, we develop a differentiable chemical-physics framework for modeling chemical mixtures, DiffMix, where geometric deep learning (GDL) is leveraged to map from molecular species, compositions and environment conditions, to physical coefficients in the mixture physics laws. In particular, we extend mixture thermodynamic and transport laws by creating learnable physical coefficients, where we use graph neural networks as the molecule encoder and enforce component-wise permutation-invariance. We start our model evaluations with thermodynamics of binary mixtures, and further benchmarked multicomponent electrolyte mixtures on their transport properties, in order to test the model generalizability. We show improved prediction accuracy and model robustness of DiffMix than its purely data-driven variants. Furthermore, we demonstrate the efficient optimization of electrolyte transport properties, built on the gradient obtained using DiffMix auto-differentiation. Our simulation runs are then backed up by the data generated by a robotic experimentation setup, Clio. By combining mixture physics and GDL, DiffMix expands the predictive modeling methods for chemical mixtures and provides low-cost optimization approaches in large chemical spaces.