 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFrom Knowledge to Action: Outcomes of the 2025 Large Language Model (LLM) Hackathon for Applications in Materials Science and Chemistry
May 04, 2026Large language models (LLMs) are rapidly changing how researchers in materials science and chemistry discover, organize, and act on scientific knowledge. This paper analyzes a broad set of community-developed LLM applications in an effort to identify emerging patterns in how these systems can be used across the scientific research lifecycle. We organize the projects into two complementary categories: Knowledge Infrastructure, systems that structure, retrieve, synthesize, and validate scientific information; and Action Systems, systems that execute, coordinate, or automate scientific work across computational and experimental environments. The submissions reveal a shift from single-purpose LLM tools toward integrated, multi-agent workflows that combine retrieval, reasoning, tool use, and domain-specific validation. Prominent themes include retrieval-augmented generation as grounding infrastructure, persistent structured knowledge representations, multimodal and multilingual scientific inputs, and early progress toward laboratory-integrated closed-loop systems. Together, these results suggest that LLMs are evolving from general-purpose assistants into composable infrastructure for scientific reasoning and action. This work provides a community snapshot of that transition and a practical taxonomy for understanding emerging LLM-enabled workflows in materials science and chemistry.
MOFSimBench: Evaluating Universal Machine Learning Interatomic Potentials In Metal--Organic Framework Molecular Modeling
Jul 16, 2025Universal machine learning interatomic potentials (uMLIPs) have emerged as powerful tools for accelerating atomistic simulations, offering scalable and efficient modeling with accuracy close to quantum calculations. However, their reliability and effectiveness in practical, real-world applications remain an open question. Metal-organic frameworks (MOFs) and related nanoporous materials are highly porous crystals with critical relevance in carbon capture, energy storage, and catalysis applications. Modeling nanoporous materials presents distinct challenges for uMLIPs due to their diverse chemistry, structural complexity, including porosity and coordination bonds, and the absence from existing training datasets. Here, we introduce MOFSimBench, a benchmark to evaluate uMLIPs on key materials modeling tasks for nanoporous materials, including structural optimization, molecular dynamics (MD) stability, the prediction of bulk properties, such as bulk modulus and heat capacity, and guest-host interactions. Evaluating over 20 models from various architectures on a chemically and structurally diverse materials set, we find that top-performing uMLIPs consistently outperform classical force fields and fine-tuned machine learning potentials across all tasks, demonstrating their readiness for deployment in nanoporous materials modeling. Our analysis highlights that data quality, particularly the diversity of training sets and inclusion of out-of-equilibrium conformations, plays a more critical role than model architecture in determining performance across all evaluated uMLIPs. We release our modular and extendable benchmarking framework at https://github.com/AI4ChemS/mofsim-bench, providing an open resource to guide the adoption for nanoporous materials modeling and further development of uMLIPs.
CheMixHub: Datasets and Benchmarks for Chemical Mixture Property Prediction
Jun 13, 2025Developing improved predictive models for multi-molecular systems is crucial, as nearly every chemical product used results from a mixture of chemicals. While being a vital part of the industry pipeline, the chemical mixture space remains relatively unexplored by the Machine Learning community. In this paper, we introduce CheMixHub, a holistic benchmark for molecular mixtures, covering a corpus of 11 chemical mixtures property prediction tasks, from drug delivery formulations to battery electrolytes, totalling approximately 500k data points gathered and curated from 7 publicly available datasets. CheMixHub introduces various data splitting techniques to assess context-specific generalization and model robustness, providing a foundation for the development of predictive models for chemical mixture properties. Furthermore, we map out the modelling space of deep learning models for chemical mixtures, establishing initial benchmarks for the community. This dataset has the potential to accelerate chemical mixture development, encompassing reformulation, optimization, and discovery. The dataset and code for the benchmarks can be found at: https://github.com/chemcognition-lab/chemixhub
34 Examples of LLM Applications in Materials Science and Chemistry: Towards Automation, Assistants, Agents, and Accelerated Scientific Discovery
May 05, 2025
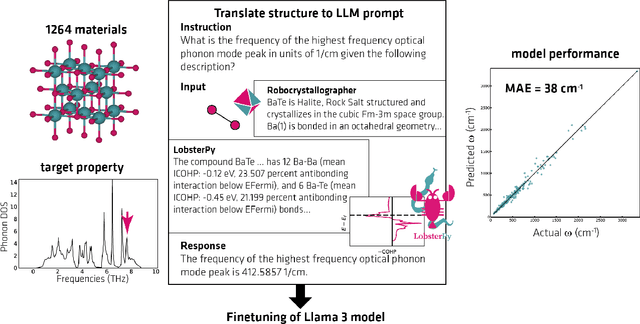
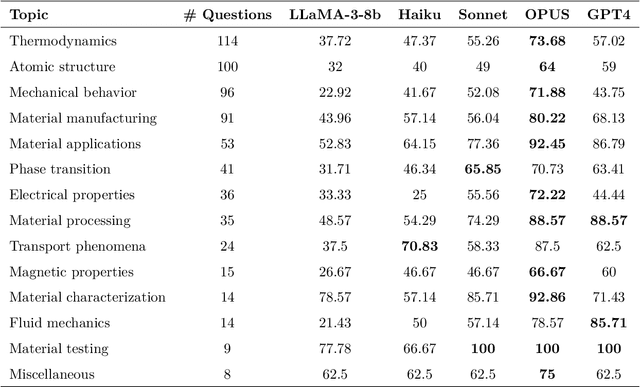
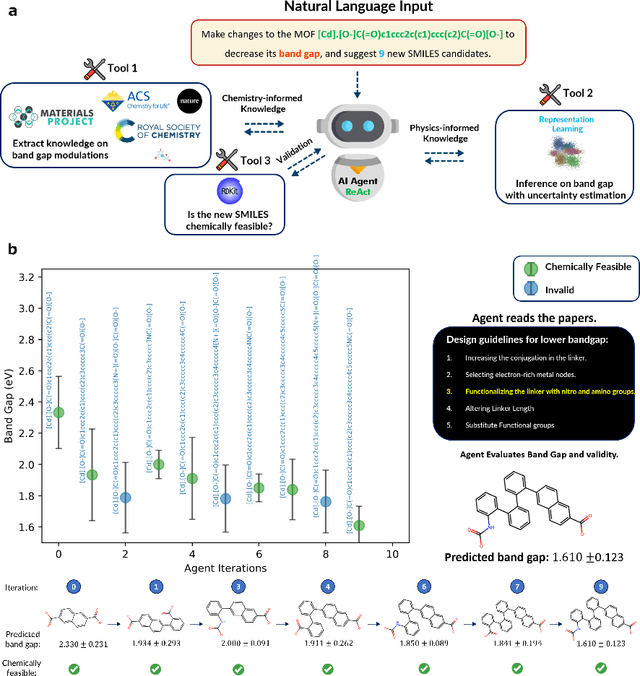
Large Language Models (LLMs) are reshaping many aspects of materials science and chemistry research, enabling advances in molecular property prediction, materials design, scientific automation, knowledge extraction, and more. Recent developments demonstrate that the latest class of models are able to integrate structured and unstructured data, assist in hypothesis generation, and streamline research workflows. To explore the frontier of LLM capabilities across the research lifecycle, we review applications of LLMs through 34 total projects developed during the second annual Large Language Model Hackathon for Applications in Materials Science and Chemistry, a global hybrid event. These projects spanned seven key research areas: (1) molecular and material property prediction, (2) molecular and material design, (3) automation and novel interfaces, (4) scientific communication and education, (5) research data management and automation, (6) hypothesis generation and evaluation, and (7) knowledge extraction and reasoning from the scientific literature. Collectively, these applications illustrate how LLMs serve as versatile predictive models, platforms for rapid prototyping of domain-specific tools, and much more. In particular, improvements in both open source and proprietary LLM performance through the addition of reasoning, additional training data, and new techniques have expanded effectiveness, particularly in low-data environments and interdisciplinary research. As LLMs continue to improve, their integration into scientific workflows presents both new opportunities and new challenges, requiring ongoing exploration, continued refinement, and further research to address reliability, interpretability, and reproducibility.
Reflections from the 2024 Large Language Model (LLM) Hackathon for Applications in Materials Science and Chemistry
Nov 20, 2024
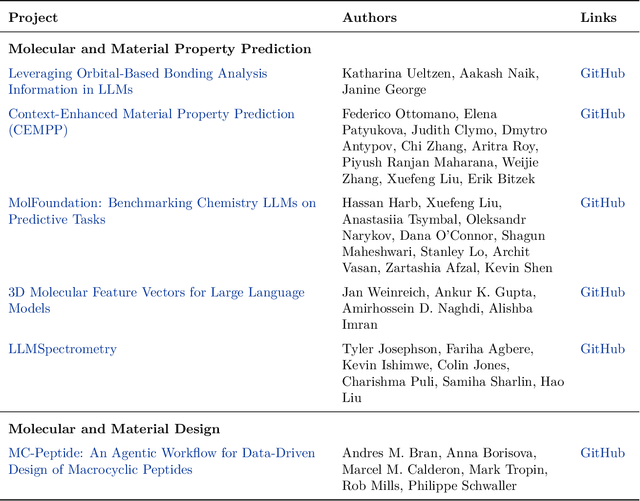
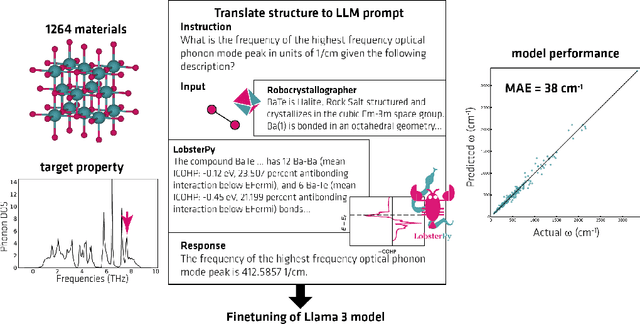
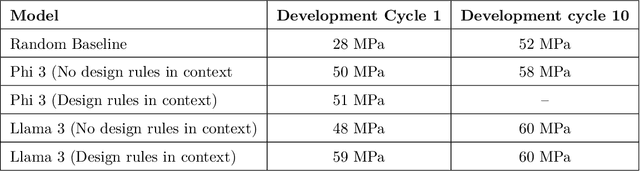
Here, we present the outcomes from the second Large Language Model (LLM) Hackathon for Applications in Materials Science and Chemistry, which engaged participants across global hybrid locations, resulting in 34 team submissions. The submissions spanned seven key application areas and demonstrated the diverse utility of LLMs for applications in (1) molecular and material property prediction; (2) molecular and material design; (3) automation and novel interfaces; (4) scientific communication and education; (5) research data management and automation; (6) hypothesis generation and evaluation; and (7) knowledge extraction and reasoning from scientific literature. Each team submission is presented in a summary table with links to the code and as brief papers in the appendix. Beyond team results, we discuss the hackathon event and its hybrid format, which included physical hubs in Toronto, Montreal, San Francisco, Berlin, Lausanne, and Tokyo, alongside a global online hub to enable local and virtual collaboration. Overall, the event highlighted significant improvements in LLM capabilities since the previous year's hackathon, suggesting continued expansion of LLMs for applications in materials science and chemistry research. These outcomes demonstrate the dual utility of LLMs as both multipurpose models for diverse machine learning tasks and platforms for rapid prototyping custom applications in scientific research.
Agent-based Learning of Materials Datasets from Scientific Literature
Dec 18, 2023Advancements in machine learning and artificial intelligence are transforming materials discovery. Yet, the availability of structured experimental data remains a bottleneck. The vast corpus of scientific literature presents a valuable and rich resource of such data. However, manual dataset creation from these resources is challenging due to issues in maintaining quality and consistency, scalability limitations, and the risk of human error and bias. Therefore, in this work, we develop a chemist AI agent, powered by large language models (LLMs), to overcome these challenges by autonomously creating structured datasets from natural language text, ranging from sentences and paragraphs to extensive scientific research articles. Our chemist AI agent, Eunomia, can plan and execute actions by leveraging the existing knowledge from decades of scientific research articles, scientists, the Internet and other tools altogether. We benchmark the performance of our approach in three different information extraction tasks with various levels of complexity, including solid-state impurity doping, metal-organic framework (MOF) chemical formula, and property relations. Our results demonstrate that our zero-shot agent, with the appropriate tools, is capable of attaining performance that is either superior or comparable to the state-of-the-art fine-tuned materials information extraction methods. This approach simplifies compilation of machine learning-ready datasets for various materials discovery applications, and significantly ease the accessibility of advanced natural language processing tools for novice users in natural language. The methodology in this work is developed as an open-source software on https://github.com/AI4ChemS/Eunomia.
SELFIES and the future of molecular string representations
Mar 31, 2022
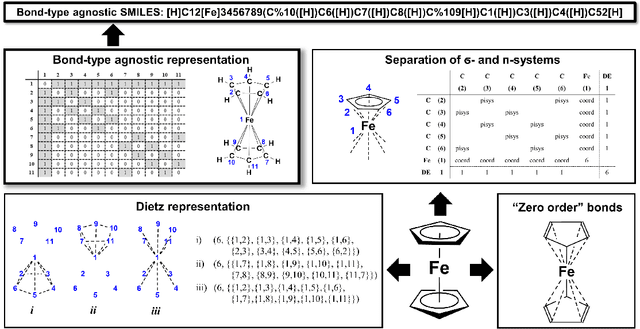
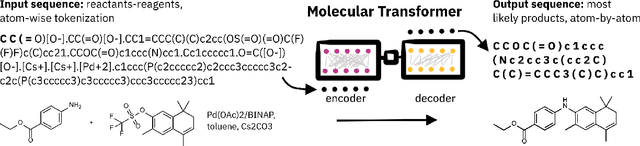
Artificial intelligence (AI) and machine learning (ML) are expanding in popularity for broad applications to challenging tasks in chemistry and materials science. Examples include the prediction of properties, the discovery of new reaction pathways, or the design of new molecules. The machine needs to read and write fluently in a chemical language for each of these tasks. Strings are a common tool to represent molecular graphs, and the most popular molecular string representation, SMILES, has powered cheminformatics since the late 1980s. However, in the context of AI and ML in chemistry, SMILES has several shortcomings -- most pertinently, most combinations of symbols lead to invalid results with no valid chemical interpretation. To overcome this issue, a new language for molecules was introduced in 2020 that guarantees 100\% robustness: SELFIES (SELF-referencIng Embedded Strings). SELFIES has since simplified and enabled numerous new applications in chemistry. In this manuscript, we look to the future and discuss molecular string representations, along with their respective opportunities and challenges. We propose 16 concrete Future Projects for robust molecular representations. These involve the extension toward new chemical domains, exciting questions at the interface of AI and robust languages and interpretability for both humans and machines. We hope that these proposals will inspire several follow-up works exploiting the full potential of molecular string representations for the future of AI in chemistry and materials science.
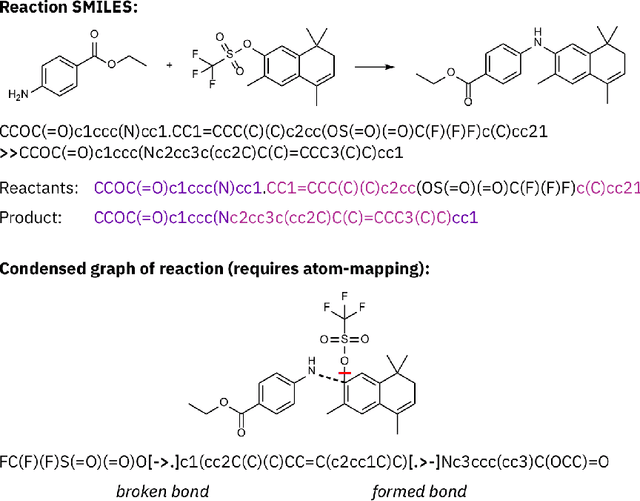
Big-Data Science in Porous Materials: Materials Genomics and Machine Learning
Jan 18, 2020
By combining metal nodes with organic linkers we can potentially synthesize millions of possible metal organic frameworks (MOFs). At present, we have libraries of over ten thousand synthesized materials and millions of in-silico predicted materials. The fact that we have so many materials opens many exciting avenues to tailor make a material that is optimal for a given application. However, from an experimental and computational point of view we simply have too many materials to screen using brute-force techniques. In this review, we show that having so many materials allows us to use big-data methods as a powerful technique to study these materials and to discover complex correlations. The first part of the review gives an introduction to the principles of big-data science. We emphasize the importance of data collection, methods to augment small data sets, how to select appropriate training sets. An important part of this review are the different approaches that are used to represent these materials in feature space. The review also includes a general overview of the different ML techniques, but as most applications in porous materials use supervised ML our review is focused on the different approaches for supervised ML. In particular, we review the different method to optimize the ML process and how to quantify the performance of the different methods. In the second part, we review how the different approaches of ML have been applied to porous materials. In particular, we discuss applications in the field of gas storage and separation, the stability of these materials, their electronic properties, and their synthesis. The range of topics illustrates the large variety of topics that can be studied with big-data science. Given the increasing interest of the scientific community in ML, we expect this list to rapidly expand in the coming years.