 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to Edge14 Examples of How LLMs Can Transform Materials Science and Chemistry: A Reflection on a Large Language Model Hackathon
Jun 13, 2023



Chemistry and materials science are complex. Recently, there have been great successes in addressing this complexity using data-driven or computational techniques. Yet, the necessity of input structured in very specific forms and the fact that there is an ever-growing number of tools creates usability and accessibility challenges. Coupled with the reality that much data in these disciplines is unstructured, the effectiveness of these tools is limited. Motivated by recent works that indicated that large language models (LLMs) might help address some of these issues, we organized a hackathon event on the applications of LLMs in chemistry, materials science, and beyond. This article chronicles the projects built as part of this hackathon. Participants employed LLMs for various applications, including predicting properties of molecules and materials, designing novel interfaces for tools, extracting knowledge from unstructured data, and developing new educational applications. The diverse topics and the fact that working prototypes could be generated in less than two days highlight that LLMs will profoundly impact the future of our fields. The rich collection of ideas and projects also indicates that the applications of LLMs are not limited to materials science and chemistry but offer potential benefits to a wide range of scientific disciplines.
SELFIES and the future of molecular string representations
Mar 31, 2022
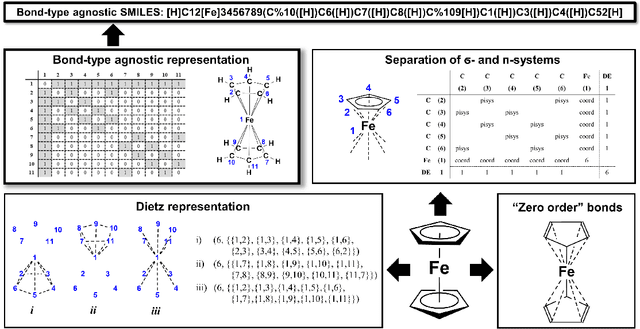
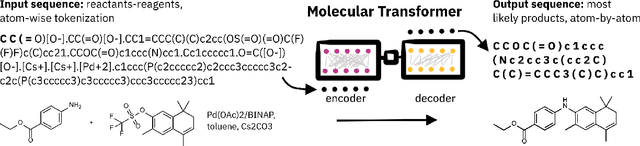
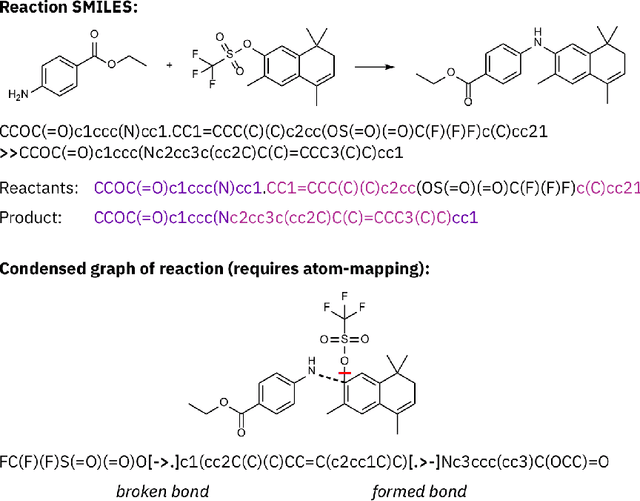
Artificial intelligence (AI) and machine learning (ML) are expanding in popularity for broad applications to challenging tasks in chemistry and materials science. Examples include the prediction of properties, the discovery of new reaction pathways, or the design of new molecules. The machine needs to read and write fluently in a chemical language for each of these tasks. Strings are a common tool to represent molecular graphs, and the most popular molecular string representation, SMILES, has powered cheminformatics since the late 1980s. However, in the context of AI and ML in chemistry, SMILES has several shortcomings -- most pertinently, most combinations of symbols lead to invalid results with no valid chemical interpretation. To overcome this issue, a new language for molecules was introduced in 2020 that guarantees 100\% robustness: SELFIES (SELF-referencIng Embedded Strings). SELFIES has since simplified and enabled numerous new applications in chemistry. In this manuscript, we look to the future and discuss molecular string representations, along with their respective opportunities and challenges. We propose 16 concrete Future Projects for robust molecular representations. These involve the extension toward new chemical domains, exciting questions at the interface of AI and robust languages and interpretability for both humans and machines. We hope that these proposals will inspire several follow-up works exploiting the full potential of molecular string representations for the future of AI in chemistry and materials science.
A reproducibility study of "Augmenting Genetic Algorithms with Deep Neural Networks for Exploring the Chemical Space"
Feb 10, 2021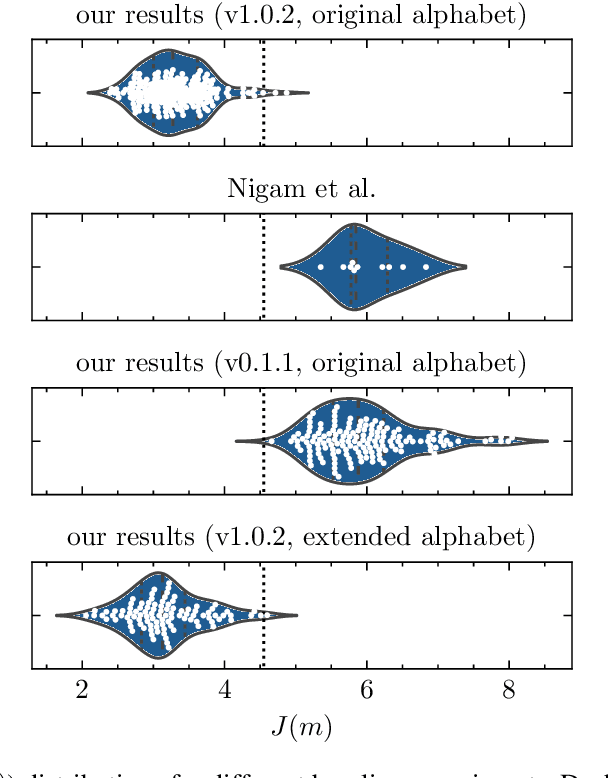

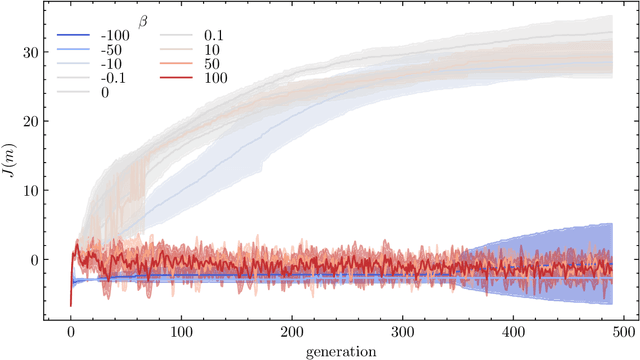
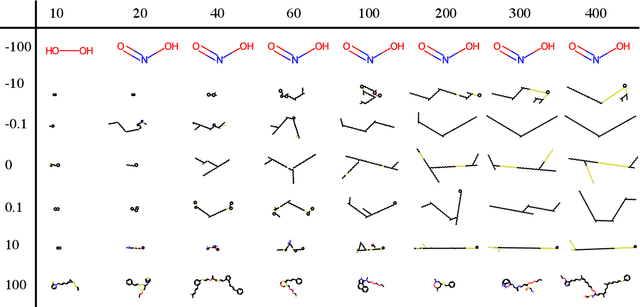
Nigam et al. reported a genetic algorithm (GA) utilizing the SELFIES representation and also propose an adaptive, neural network-based penalty that is supposed to improve the diversity of the generated molecules. The main claims of the paper are that this GA outperforms other generative techniques (as measured by the penalized logP) and that a neural network-based adaptive penalty increases the diversity of the generated molecules. In this work, we investigated the reproducibility of their claims. Overall, we were able to reproduce comparable results using the SELFIES-based GA, but mostly by exploiting deficiencies of the (easily optimizable) fitness function (i.e., generating long, sulfur containing chains). In addition, we reproduce results showing that the discriminator can be used to bias the generation of molecules to ones that are similar to the reference set. Lastly, we attempted to quantify the evolution of the diversity, understand the influence of some hyperparameters, and propose improvements to the adaptive penalty.
Big-Data Science in Porous Materials: Materials Genomics and Machine Learning
Jan 18, 2020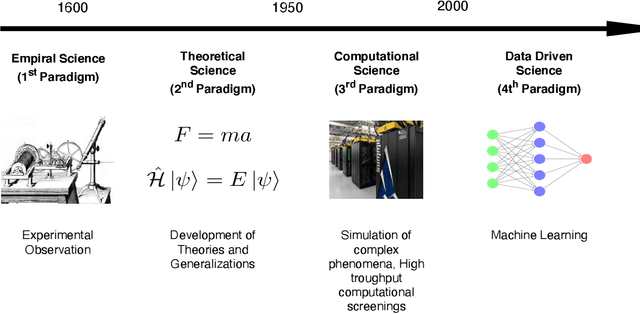
By combining metal nodes with organic linkers we can potentially synthesize millions of possible metal organic frameworks (MOFs). At present, we have libraries of over ten thousand synthesized materials and millions of in-silico predicted materials. The fact that we have so many materials opens many exciting avenues to tailor make a material that is optimal for a given application. However, from an experimental and computational point of view we simply have too many materials to screen using brute-force techniques. In this review, we show that having so many materials allows us to use big-data methods as a powerful technique to study these materials and to discover complex correlations. The first part of the review gives an introduction to the principles of big-data science. We emphasize the importance of data collection, methods to augment small data sets, how to select appropriate training sets. An important part of this review are the different approaches that are used to represent these materials in feature space. The review also includes a general overview of the different ML techniques, but as most applications in porous materials use supervised ML our review is focused on the different approaches for supervised ML. In particular, we review the different method to optimize the ML process and how to quantify the performance of the different methods. In the second part, we review how the different approaches of ML have been applied to porous materials. In particular, we discuss applications in the field of gas storage and separation, the stability of these materials, their electronic properties, and their synthesis. The range of topics illustrates the large variety of topics that can be studied with big-data science. Given the increasing interest of the scientific community in ML, we expect this list to rapidly expand in the coming years.