 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDisCo-Diff: Enhancing Continuous Diffusion Models with Discrete Latents
Jul 03, 2024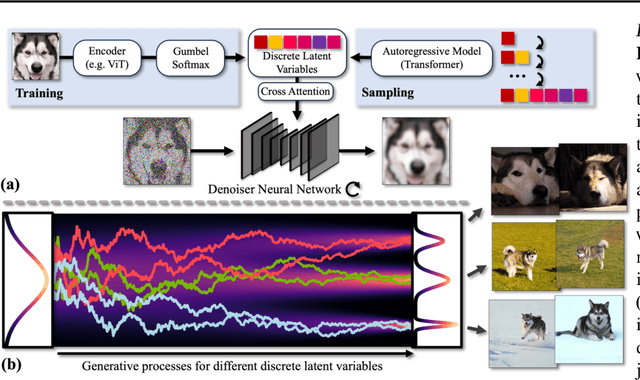
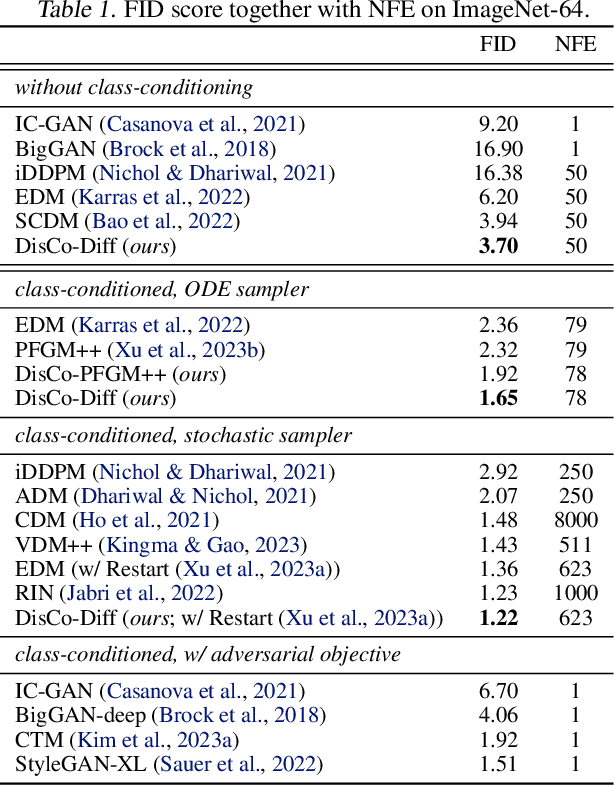

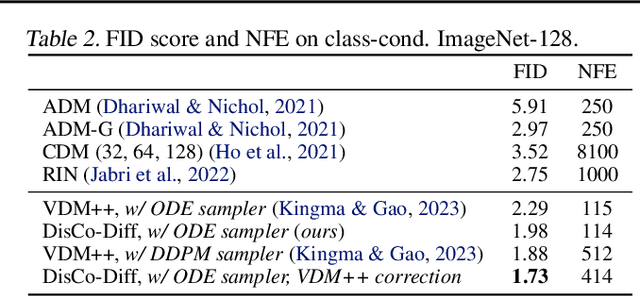
Diffusion models (DMs) have revolutionized generative learning. They utilize a diffusion process to encode data into a simple Gaussian distribution. However, encoding a complex, potentially multimodal data distribution into a single continuous Gaussian distribution arguably represents an unnecessarily challenging learning problem. We propose Discrete-Continuous Latent Variable Diffusion Models (DisCo-Diff) to simplify this task by introducing complementary discrete latent variables. We augment DMs with learnable discrete latents, inferred with an encoder, and train DM and encoder end-to-end. DisCo-Diff does not rely on pre-trained networks, making the framework universally applicable. The discrete latents significantly simplify learning the DM's complex noise-to-data mapping by reducing the curvature of the DM's generative ODE. An additional autoregressive transformer models the distribution of the discrete latents, a simple step because DisCo-Diff requires only few discrete variables with small codebooks. We validate DisCo-Diff on toy data, several image synthesis tasks as well as molecular docking, and find that introducing discrete latents consistently improves model performance. For example, DisCo-Diff achieves state-of-the-art FID scores on class-conditioned ImageNet-64/128 datasets with ODE sampler.
Deep Confident Steps to New Pockets: Strategies for Docking Generalization
Feb 28, 2024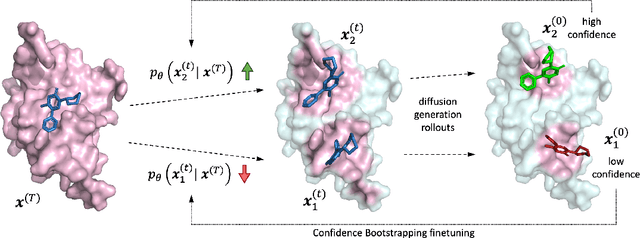
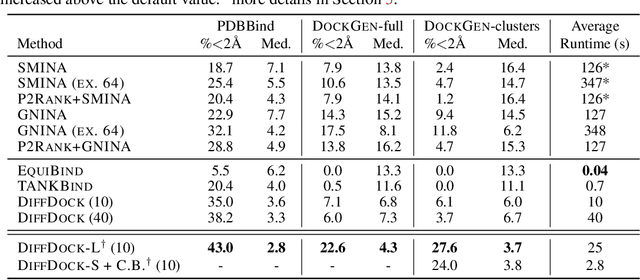
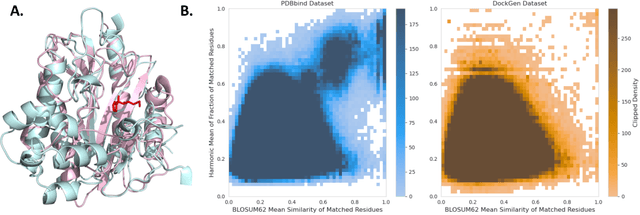
Accurate blind docking has the potential to lead to new biological breakthroughs, but for this promise to be realized, docking methods must generalize well across the proteome. Existing benchmarks, however, fail to rigorously assess generalizability. Therefore, we develop DockGen, a new benchmark based on the ligand-binding domains of proteins, and we show that existing machine learning-based docking models have very weak generalization abilities. We carefully analyze the scaling laws of ML-based docking and show that, by scaling data and model size, as well as integrating synthetic data strategies, we are able to significantly increase the generalization capacity and set new state-of-the-art performance across benchmarks. Further, we propose Confidence Bootstrapping, a new training paradigm that solely relies on the interaction between diffusion and confidence models and exploits the multi-resolution generation process of diffusion models. We demonstrate that Confidence Bootstrapping significantly improves the ability of ML-based docking methods to dock to unseen protein classes, edging closer to accurate and generalizable blind docking methods.
Dirichlet Flow Matching with Applications to DNA Sequence Design
Feb 08, 2024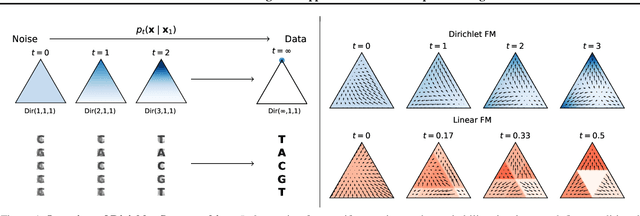

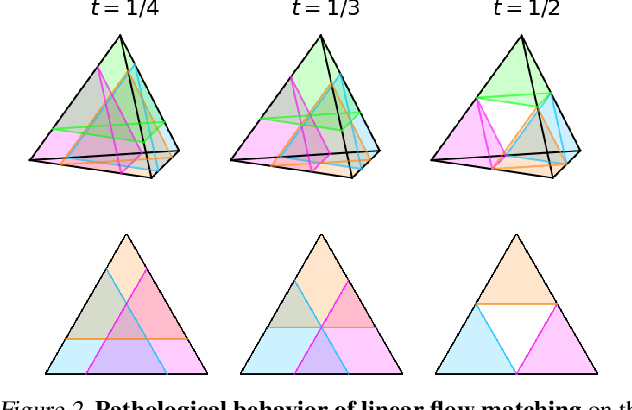
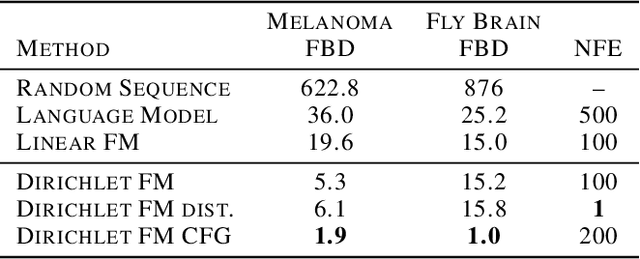
Discrete diffusion or flow models could enable faster and more controllable sequence generation than autoregressive models. We show that na\"ive linear flow matching on the simplex is insufficient toward this goal since it suffers from discontinuities in the training target and further pathologies. To overcome this, we develop Dirichlet flow matching on the simplex based on mixtures of Dirichlet distributions as probability paths. In this framework, we derive a connection between the mixtures' scores and the flow's vector field that allows for classifier and classifier-free guidance. Further, we provide distilled Dirichlet flow matching, which enables one-step sequence generation with minimal performance hits, resulting in $O(L)$ speedups compared to autoregressive models. On complex DNA sequence generation tasks, we demonstrate superior performance compared to all baselines in distributional metrics and in achieving desired design targets for generated sequences. Finally, we show that our classifier-free guidance approach improves unconditional generation and is effective for generating DNA that satisfies design targets. Code is available at https://github.com/HannesStark/dirichlet-flow-matching.
Particle Guidance: non-I.I.D. Diverse Sampling with Diffusion Models
Oct 19, 2023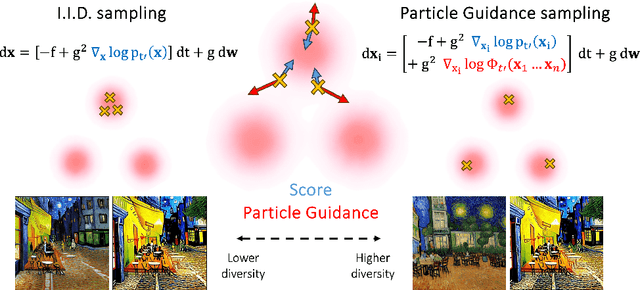
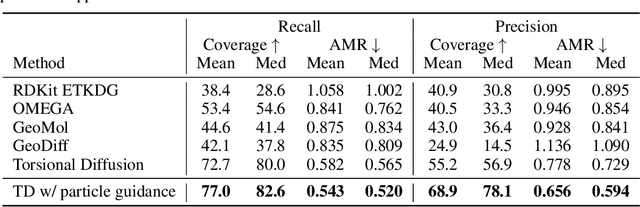
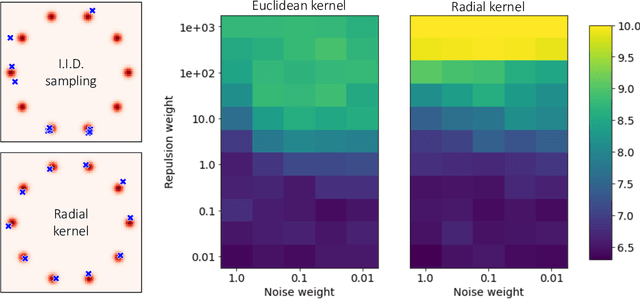
In light of the widespread success of generative models, a significant amount of research has gone into speeding up their sampling time. However, generative models are often sampled multiple times to obtain a diverse set incurring a cost that is orthogonal to sampling time. We tackle the question of how to improve diversity and sample efficiency by moving beyond the common assumption of independent samples. We propose particle guidance, an extension of diffusion-based generative sampling where a joint-particle time-evolving potential enforces diversity. We analyze theoretically the joint distribution that particle guidance generates, its implications on the choice of potential, and the connections with methods in other disciplines. Empirically, we test the framework both in the setting of conditional image generation, where we are able to increase diversity without affecting quality, and molecular conformer generation, where we reduce the state-of-the-art median error by 13% on average.
DiffDock-PP: Rigid Protein-Protein Docking with Diffusion Models
Apr 08, 2023



Understanding how proteins structurally interact is crucial to modern biology, with applications in drug discovery and protein design. Recent machine learning methods have formulated protein-small molecule docking as a generative problem with significant performance boosts over both traditional and deep learning baselines. In this work, we propose a similar approach for rigid protein-protein docking: DiffDock-PP is a diffusion generative model that learns to translate and rotate unbound protein structures into their bound conformations. We achieve state-of-the-art performance on DIPS with a median C-RMSD of 4.85, outperforming all considered baselines. Additionally, DiffDock-PP is faster than all search-based methods and generates reliable confidence estimates for its predictions. Our code is publicly available at $\texttt{https://github.com/ketatam/DiffDock-PP}$
EigenFold: Generative Protein Structure Prediction with Diffusion Models
Apr 05, 2023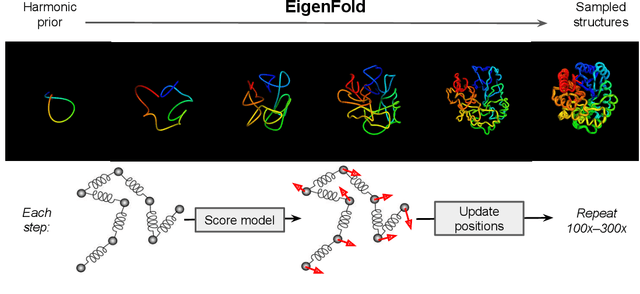
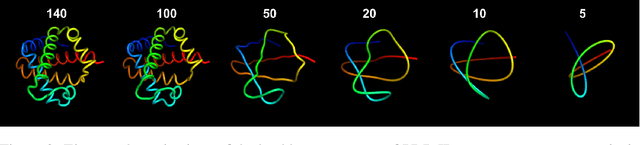
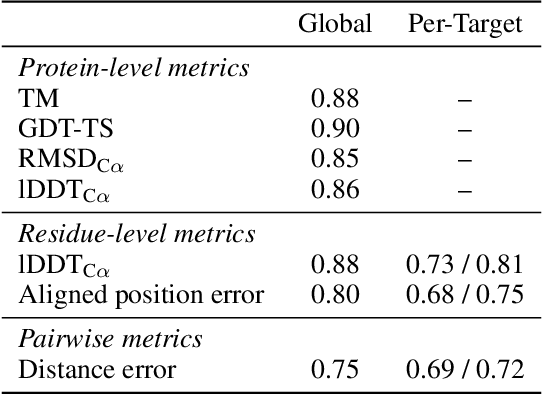
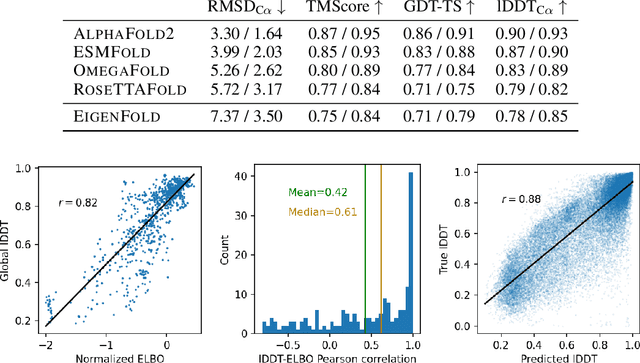
Protein structure prediction has reached revolutionary levels of accuracy on single structures, yet distributional modeling paradigms are needed to capture the conformational ensembles and flexibility that underlie biological function. Towards this goal, we develop EigenFold, a diffusion generative modeling framework for sampling a distribution of structures from a given protein sequence. We define a diffusion process that models the structure as a system of harmonic oscillators and which naturally induces a cascading-resolution generative process along the eigenmodes of the system. On recent CAMEO targets, EigenFold achieves a median TMScore of 0.84, while providing a more comprehensive picture of model uncertainty via the ensemble of sampled structures relative to existing methods. We then assess EigenFold's ability to model and predict conformational heterogeneity for fold-switching proteins and ligand-induced conformational change. Code is available at https://github.com/bjing2016/EigenFold.
Modeling Molecular Structures with Intrinsic Diffusion Models
Feb 23, 2023



Since its foundations, more than one hundred years ago, the field of structural biology has strived to understand and analyze the properties of molecules and their interactions by studying the structure that they take in 3D space. However, a fundamental challenge with this approach has been the dynamic nature of these particles, which forces us to model not a single but a whole distribution of structures for every molecular system. This thesis proposes Intrinsic Diffusion Modeling, a novel approach to this problem based on combining diffusion generative models with scientific knowledge about the flexibility of biological complexes. The knowledge of these degrees of freedom is translated into the definition of a manifold over which the diffusion process is defined. This manifold significantly reduces the dimensionality and increases the smoothness of the generation space allowing for significantly faster and more accurate generative processes. We demonstrate the effectiveness of this approach on two fundamental tasks at the basis of computational chemistry and biology: molecular conformer generation and molecular docking. In both tasks, we construct the first deep learning method to outperform traditional computational approaches achieving an unprecedented level of accuracy for scalable programs.
Learning Graph Search Heuristics
Dec 07, 2022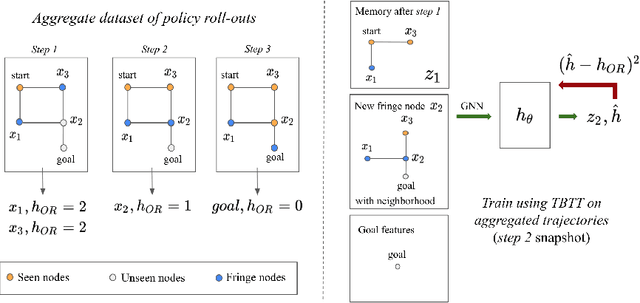
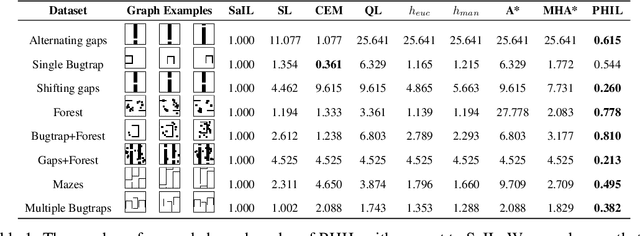
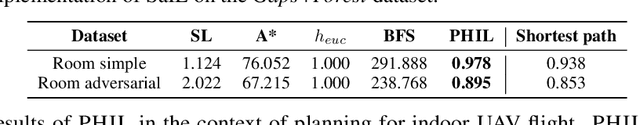
Searching for a path between two nodes in a graph is one of the most well-studied and fundamental problems in computer science. In numerous domains such as robotics, AI, or biology, practitioners develop search heuristics to accelerate their pathfinding algorithms. However, it is a laborious and complex process to hand-design heuristics based on the problem and the structure of a given use case. Here we present PHIL (Path Heuristic with Imitation Learning), a novel neural architecture and a training algorithm for discovering graph search and navigation heuristics from data by leveraging recent advances in imitation learning and graph representation learning. At training time, we aggregate datasets of search trajectories and ground-truth shortest path distances, which we use to train a specialized graph neural network-based heuristic function using backpropagation through steps of the pathfinding process. Our heuristic function learns graph embeddings useful for inferring node distances, runs in constant time independent of graph sizes, and can be easily incorporated in an algorithm such as A* at test time. Experiments show that PHIL reduces the number of explored nodes compared to state-of-the-art methods on benchmark datasets by 58.5\% on average, can be directly applied in diverse graphs ranging from biological networks to road networks, and allows for fast planning in time-critical robotics domains.
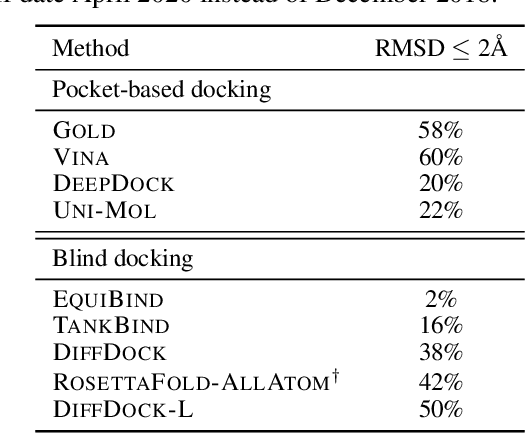
DiffDock: Diffusion Steps, Twists, and Turns for Molecular Docking
Oct 04, 2022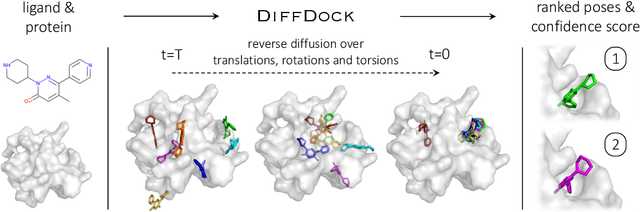
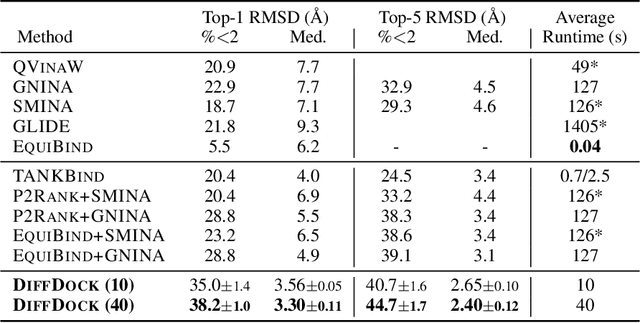
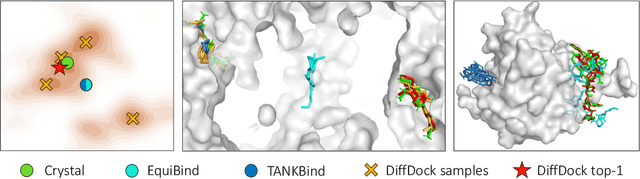

Predicting the binding structure of a small molecule ligand to a protein -- a task known as molecular docking -- is critical to drug design. Recent deep learning methods that treat docking as a regression problem have decreased runtime compared to traditional search-based methods but have yet to offer substantial improvements in accuracy. We instead frame molecular docking as a generative modeling problem and develop DiffDock, a diffusion generative model over the non-Euclidean manifold of ligand poses. To do so, we map this manifold to the product space of the degrees of freedom (translational, rotational, and torsional) involved in docking and develop an efficient diffusion process on this space. Empirically, DiffDock obtains a 38% top-1 success rate (RMSD<2A) on PDBBind, significantly outperforming the previous state-of-the-art of traditional docking (23%) and deep learning (20%) methods. Moreover, DiffDock has fast inference times and provides confidence estimates with high selective accuracy.
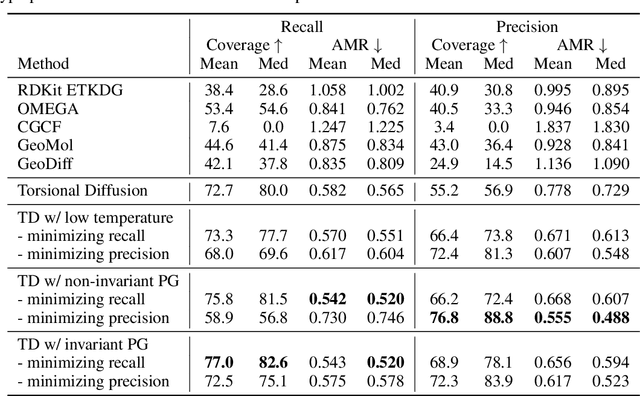
Torsional Diffusion for Molecular Conformer Generation
Jun 01, 2022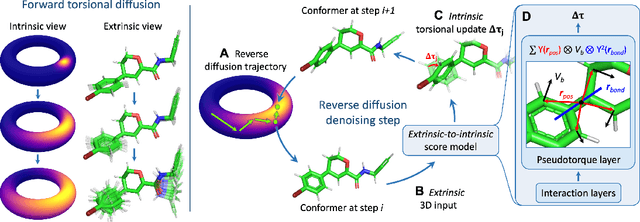
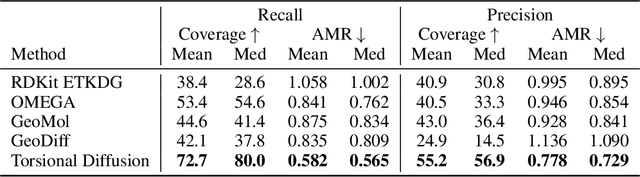
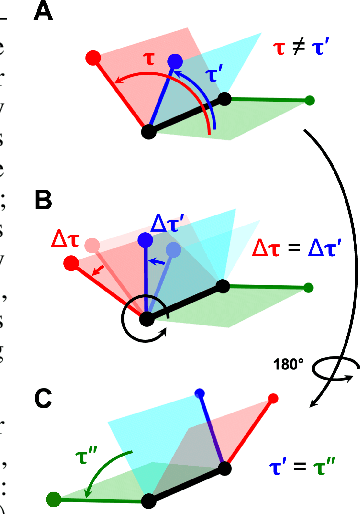
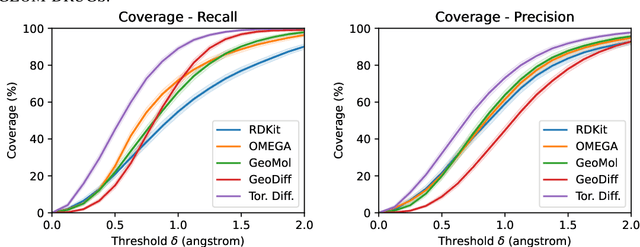
Molecular conformer generation is a fundamental task in computational chemistry. Several machine learning approaches have been developed, but none have outperformed state-of-the-art cheminformatics methods. We propose torsional diffusion, a novel diffusion framework that operates on the space of torsion angles via a diffusion process on the hypertorus and an extrinsic-to-intrinsic score model. On a standard benchmark of drug-like molecules, torsional diffusion generates superior conformer ensembles compared to machine learning and cheminformatics methods in terms of both RMSD and chemical properties, and is orders of magnitude faster than previous diffusion-based models. Moreover, our model provides exact likelihoods, which we employ to build the first generalizable Boltzmann generator. Code is available at https://github.com/gcorso/torsional-diffusion.