 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Bayesian Model for Multi-stage Censoring
Nov 18, 2025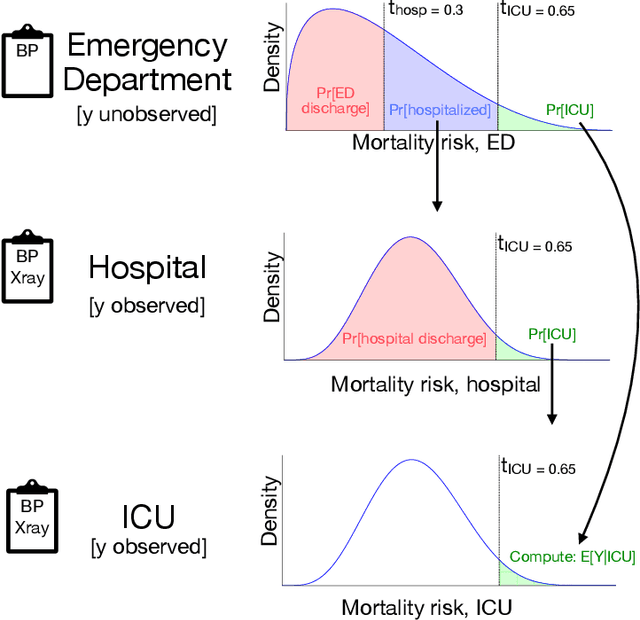
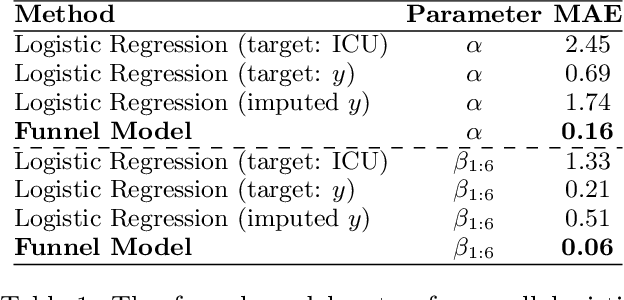

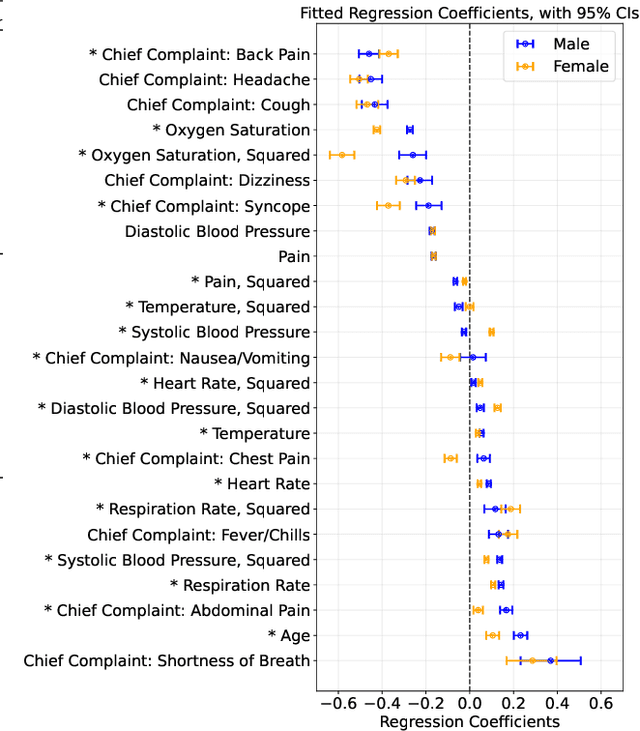
Many sequential decision settings in healthcare feature funnel structures characterized by a series of stages, such as screenings or evaluations, where the number of patients who advance to each stage progressively decreases and decisions become increasingly costly. For example, an oncologist may first conduct a breast exam, followed by a mammogram for patients with concerning exams, followed by a biopsy for patients with concerning mammograms. A key challenge is that the ground truth outcome, such as the biopsy result, is only revealed at the end of this funnel. The selective censoring of the ground truth can introduce statistical biases in risk estimation, especially in underserved patient groups, whose outcomes are more frequently censored. We develop a Bayesian model for funnel decision structures, drawing from prior work on selective labels and censoring. We first show in synthetic settings that our model is able to recover the true parameters and predict outcomes for censored patients more accurately than baselines. We then apply our model to a dataset of emergency department visits, where in-hospital mortality is observed only for those who are admitted to either the hospital or ICU. We find that there are gender-based differences in hospital and ICU admissions. In particular, our model estimates that the mortality risk threshold to admit women to the ICU is higher for women (5.1%) than for men (4.5%).
Urban Incident Prediction with Graph Neural Networks: Integrating Government Ratings and Crowdsourced Reports
Jun 10, 2025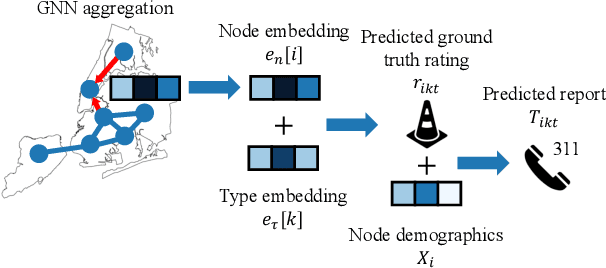

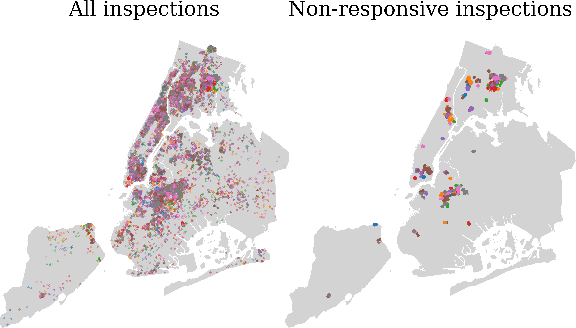
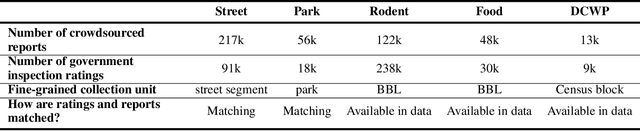
Graph neural networks (GNNs) are widely used in urban spatiotemporal forecasting, such as predicting infrastructure problems. In this setting, government officials wish to know in which neighborhoods incidents like potholes or rodent issues occur. The true state of incidents (e.g., street conditions) for each neighborhood is observed via government inspection ratings. However, these ratings are only conducted for a sparse set of neighborhoods and incident types. We also observe the state of incidents via crowdsourced reports, which are more densely observed but may be biased due to heterogeneous reporting behavior. First, for such settings, we propose a multiview, multioutput GNN-based model that uses both unbiased rating data and biased reporting data to predict the true latent state of incidents. Second, we investigate a case study of New York City urban incidents and collect, standardize, and make publicly available a dataset of 9,615,863 crowdsourced reports and 1,041,415 government inspection ratings over 3 years and across 139 types of incidents. Finally, we show on both real and semi-synthetic data that our model can better predict the latent state compared to models that use only reporting data or models that use only rating data, especially when rating data is sparse and reports are predictive of ratings. We also quantify demographic biases in crowdsourced reporting, e.g., higher-income neighborhoods report problems at higher rates. Our analysis showcases a widely applicable approach for latent state prediction using heterogeneous, sparse, and biased data.
Evaluating multiple models using labeled and unlabeled data
Jan 21, 2025



It remains difficult to evaluate machine learning classifiers in the absence of a large, labeled dataset. While labeled data can be prohibitively expensive or impossible to obtain, unlabeled data is plentiful. Here, we introduce Semi-Supervised Model Evaluation (SSME), a method that uses both labeled and unlabeled data to evaluate machine learning classifiers. SSME is the first evaluation method to take advantage of the fact that: (i) there are frequently multiple classifiers for the same task, (ii) continuous classifier scores are often available for all classes, and (iii) unlabeled data is often far more plentiful than labeled data. The key idea is to use a semi-supervised mixture model to estimate the joint distribution of ground truth labels and classifier predictions. We can then use this model to estimate any metric that is a function of classifier scores and ground truth labels (e.g., accuracy or expected calibration error). We present experiments in four domains where obtaining large labeled datasets is often impractical: (1) healthcare, (2) content moderation, (3) molecular property prediction, and (4) image annotation. Our results demonstrate that SSME estimates performance more accurately than do competing methods, reducing error by 5.1x relative to using labeled data alone and 2.4x relative to the next best competing method. SSME also improves accuracy when evaluating performance across subsets of the test distribution (e.g., specific demographic subgroups) and when evaluating the performance of language models.
Generative Modeling of Molecular Dynamics Trajectories
Sep 26, 2024



Molecular dynamics (MD) is a powerful technique for studying microscopic phenomena, but its computational cost has driven significant interest in the development of deep learning-based surrogate models. We introduce generative modeling of molecular trajectories as a paradigm for learning flexible multi-task surrogate models of MD from data. By conditioning on appropriately chosen frames of the trajectory, we show such generative models can be adapted to diverse tasks such as forward simulation, transition path sampling, and trajectory upsampling. By alternatively conditioning on part of the molecular system and inpainting the rest, we also demonstrate the first steps towards dynamics-conditioned molecular design. We validate the full set of these capabilities on tetrapeptide simulations and show that our model can produce reasonable ensembles of protein monomers. Altogether, our work illustrates how generative modeling can unlock value from MD data towards diverse downstream tasks that are not straightforward to address with existing methods or even MD itself. Code is available at https://github.com/bjing2016/mdgen.
Dirichlet Flow Matching with Applications to DNA Sequence Design
Feb 08, 2024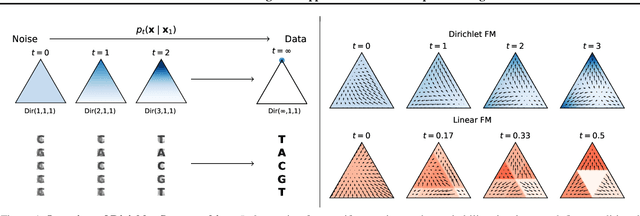

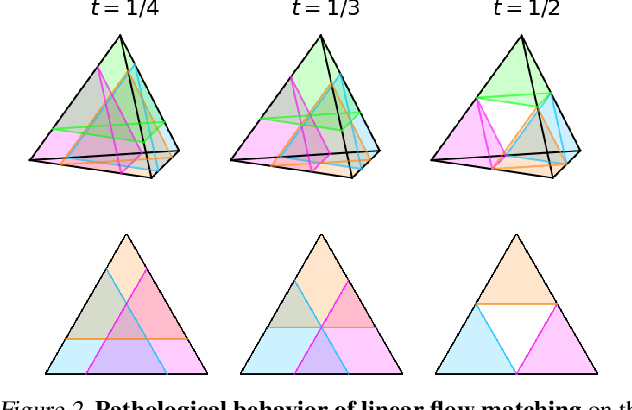
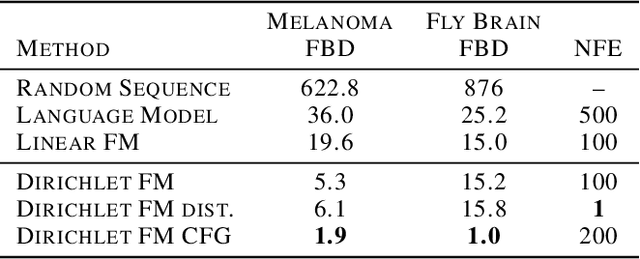
Discrete diffusion or flow models could enable faster and more controllable sequence generation than autoregressive models. We show that na\"ive linear flow matching on the simplex is insufficient toward this goal since it suffers from discontinuities in the training target and further pathologies. To overcome this, we develop Dirichlet flow matching on the simplex based on mixtures of Dirichlet distributions as probability paths. In this framework, we derive a connection between the mixtures' scores and the flow's vector field that allows for classifier and classifier-free guidance. Further, we provide distilled Dirichlet flow matching, which enables one-step sequence generation with minimal performance hits, resulting in $O(L)$ speedups compared to autoregressive models. On complex DNA sequence generation tasks, we demonstrate superior performance compared to all baselines in distributional metrics and in achieving desired design targets for generated sequences. Finally, we show that our classifier-free guidance approach improves unconditional generation and is effective for generating DNA that satisfies design targets. Code is available at https://github.com/HannesStark/dirichlet-flow-matching.
AlphaFold Meets Flow Matching for Generating Protein Ensembles
Feb 07, 2024



The biological functions of proteins often depend on dynamic structural ensembles. In this work, we develop a flow-based generative modeling approach for learning and sampling the conformational landscapes of proteins. We repurpose highly accurate single-state predictors such as AlphaFold and ESMFold and fine-tune them under a custom flow matching framework to obtain sequence-conditoned generative models of protein structure called AlphaFlow and ESMFlow. When trained and evaluated on the PDB, our method provides a superior combination of precision and diversity compared to AlphaFold with MSA subsampling. When further trained on ensembles from all-atom MD, our method accurately captures conformational flexibility, positional distributions, and higher-order ensemble observables for unseen proteins. Moreover, our method can diversify a static PDB structure with faster wall-clock convergence to certain equilibrium properties than replicate MD trajectories, demonstrating its potential as a proxy for expensive physics-based simulations. Code is available at https://github.com/bjing2016/alphaflow.
Equivariant Scalar Fields for Molecular Docking with Fast Fourier Transforms
Dec 07, 2023Molecular docking is critical to structure-based virtual screening, yet the throughput of such workflows is limited by the expensive optimization of scoring functions involved in most docking algorithms. We explore how machine learning can accelerate this process by learning a scoring function with a functional form that allows for more rapid optimization. Specifically, we define the scoring function to be the cross-correlation of multi-channel ligand and protein scalar fields parameterized by equivariant graph neural networks, enabling rapid optimization over rigid-body degrees of freedom with fast Fourier transforms. The runtime of our approach can be amortized at several levels of abstraction, and is particularly favorable for virtual screening settings with a common binding pocket. We benchmark our scoring functions on two simplified docking-related tasks: decoy pose scoring and rigid conformer docking. Our method attains similar but faster performance on crystal structures compared to the widely-used Vina and Gnina scoring functions, and is more robust on computationally predicted structures. Code is available at https://github.com/bjing2016/scalar-fields.
EigenFold: Generative Protein Structure Prediction with Diffusion Models
Apr 05, 2023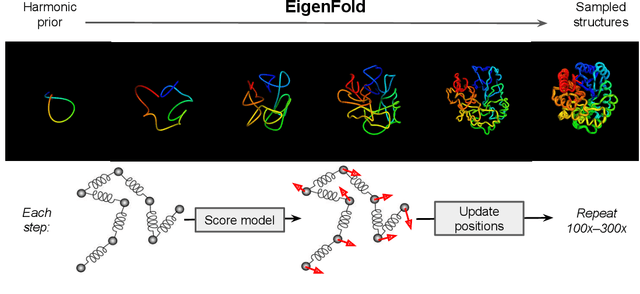
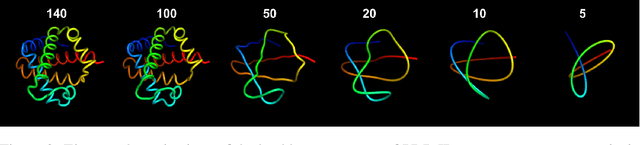
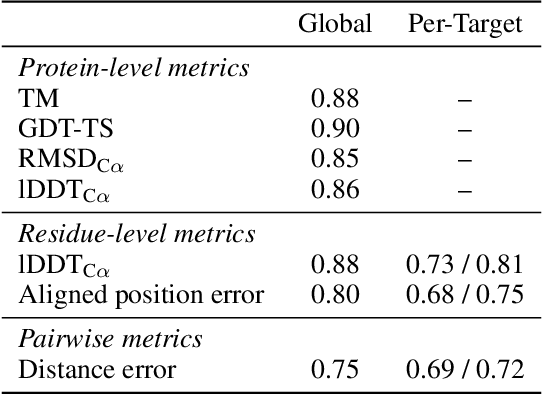
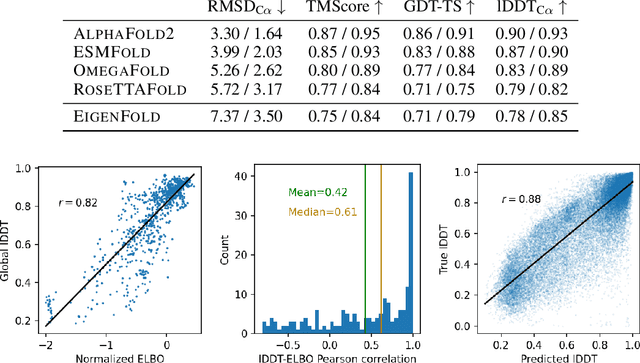
Protein structure prediction has reached revolutionary levels of accuracy on single structures, yet distributional modeling paradigms are needed to capture the conformational ensembles and flexibility that underlie biological function. Towards this goal, we develop EigenFold, a diffusion generative modeling framework for sampling a distribution of structures from a given protein sequence. We define a diffusion process that models the structure as a system of harmonic oscillators and which naturally induces a cascading-resolution generative process along the eigenmodes of the system. On recent CAMEO targets, EigenFold achieves a median TMScore of 0.84, while providing a more comprehensive picture of model uncertainty via the ensemble of sampled structures relative to existing methods. We then assess EigenFold's ability to model and predict conformational heterogeneity for fold-switching proteins and ligand-induced conformational change. Code is available at https://github.com/bjing2016/EigenFold.
Granger causal inference on DAGs identifies genomic loci regulating transcription
Oct 18, 2022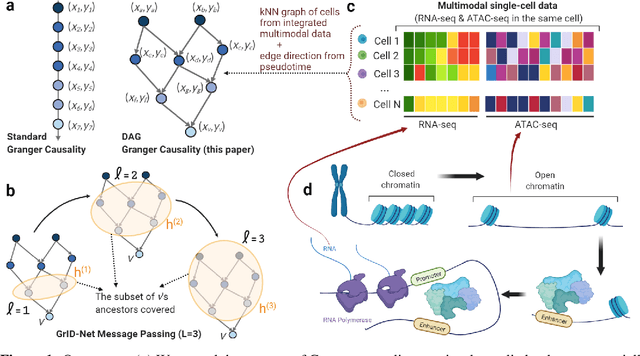
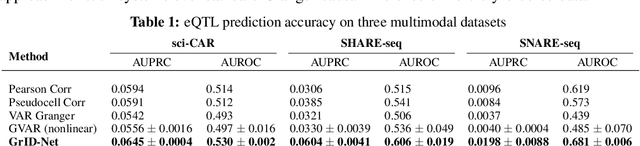
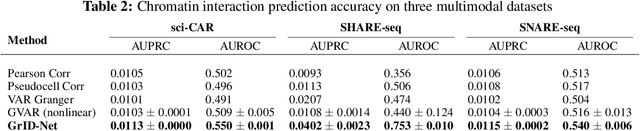
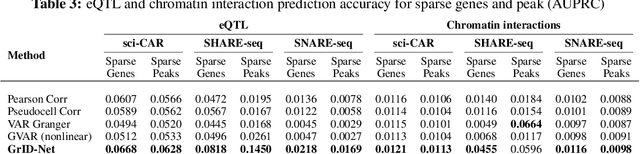
When a dynamical system can be modeled as a sequence of observations, Granger causality is a powerful approach for detecting predictive interactions between its variables. However, traditional Granger causal inference has limited utility in domains where the dynamics need to be represented as directed acyclic graphs (DAGs) rather than as a linear sequence, such as with cell differentiation trajectories. Here, we present GrID-Net, a framework based on graph neural networks with lagged message passing for Granger causal inference on DAG-structured systems. Our motivating application is the analysis of single-cell multimodal data to identify genomic loci that mediate the regulation of specific genes. To our knowledge, GrID-Net is the first single-cell analysis tool that accounts for the temporal lag between a genomic locus becoming accessible and its downstream effect on a target gene's expression. We applied GrID-Net on multimodal single-cell assays that profile chromatin accessibility (ATAC-seq) and gene expression (RNA-seq) in the same cell and show that it dramatically outperforms existing methods for inferring regulatory locus-gene links, achieving up to 71% greater agreement with independent population genetics-based estimates. By extending Granger causality to DAG-structured dynamical systems, our work unlocks new domains for causal analyses and, more specifically, opens a path towards elucidating gene regulatory interactions relevant to cellular differentiation and complex human diseases at unprecedented scale and resolution.
Explicitly disentangling image content from translation and rotation with spatial-VAE
Sep 25, 2019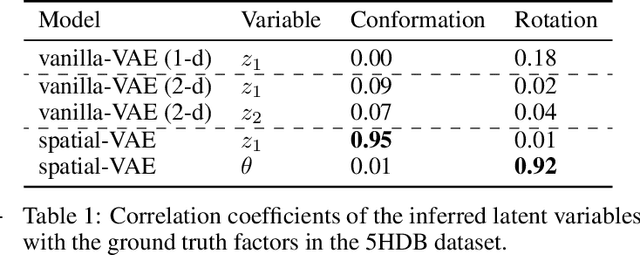
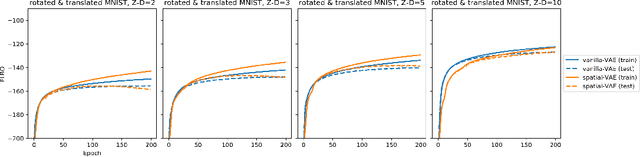

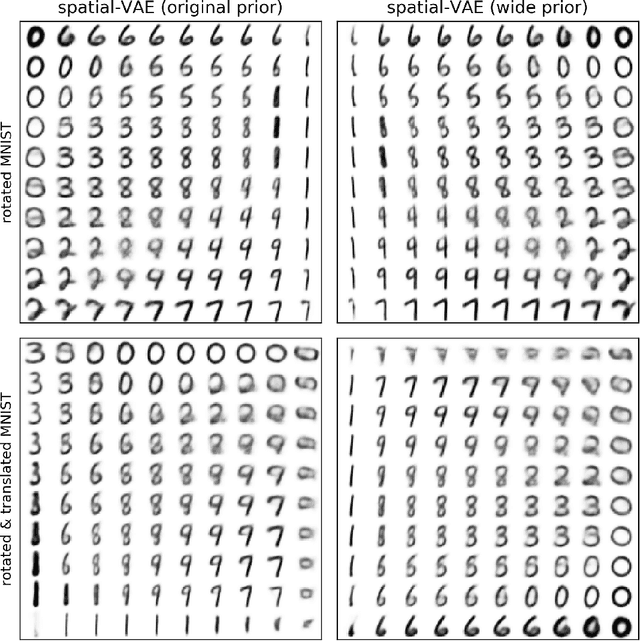
Given an image dataset, we are often interested in finding data generative factors that encode semantic content independently from pose variables such as rotation and translation. However, current disentanglement approaches do not impose any specific structure on the learned latent representations. We propose a method for explicitly disentangling image rotation and translation from other unstructured latent factors in a variational autoencoder (VAE) framework. By formulating the generative model as a function of the spatial coordinate, we make the reconstruction error differentiable with respect to latent translation and rotation parameters. This formulation allows us to train a neural network to perform approximate inference on these latent variables while explicitly constraining them to only represent rotation and translation. We demonstrate that this framework, termed spatial-VAE, effectively learns latent representations that disentangle image rotation and translation from content and improves reconstruction over standard VAEs on several benchmark datasets, including applications to modeling continuous 2-D views of proteins from single particle electron microscopy and galaxies in astronomical images.