 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGenerative Adversarial Networks Bridging Art and Machine Intelligence
Feb 09, 2025



Generative Adversarial Networks (GAN) have greatly influenced the development of computer vision and artificial intelligence in the past decade and also connected art and machine intelligence together. This book begins with a detailed introduction to the fundamental principles and historical development of GANs, contrasting them with traditional generative models and elucidating the core adversarial mechanisms through illustrative Python examples. The text systematically addresses the mathematical and theoretical underpinnings including probability theory, statistics, and game theory providing a solid framework for understanding the objectives, loss functions, and optimisation challenges inherent to GAN training. Subsequent chapters review classic variants such as Conditional GANs, DCGANs, InfoGAN, and LAPGAN before progressing to advanced training methodologies like Wasserstein GANs, GANs with gradient penalty, least squares GANs, and spectral normalisation techniques. The book further examines architectural enhancements and task-specific adaptations in generators and discriminators, showcasing practical implementations in high resolution image generation, artistic style transfer, video synthesis, text to image generation and other multimedia applications. The concluding sections offer insights into emerging research trends, including self-attention mechanisms, transformer-based generative models, and a comparative analysis with diffusion models, thus charting promising directions for future developments in both academic and applied settings.
Deep Learning Model Security: Threats and Defenses
Dec 12, 2024
Deep learning has transformed AI applications but faces critical security challenges, including adversarial attacks, data poisoning, model theft, and privacy leakage. This survey examines these vulnerabilities, detailing their mechanisms and impact on model integrity and confidentiality. Practical implementations, including adversarial examples, label flipping, and backdoor attacks, are explored alongside defenses such as adversarial training, differential privacy, and federated learning, highlighting their strengths and limitations. Advanced methods like contrastive and self-supervised learning are presented for enhancing robustness. The survey concludes with future directions, emphasizing automated defenses, zero-trust architectures, and the security challenges of large AI models. A balanced approach to performance and security is essential for developing reliable deep learning systems.
Deep Learning, Machine Learning, Advancing Big Data Analytics and Management
Dec 03, 2024



Advancements in artificial intelligence, machine learning, and deep learning have catalyzed the transformation of big data analytics and management into pivotal domains for research and application. This work explores the theoretical foundations, methodological advancements, and practical implementations of these technologies, emphasizing their role in uncovering actionable insights from massive, high-dimensional datasets. The study presents a systematic overview of data preprocessing techniques, including data cleaning, normalization, integration, and dimensionality reduction, to prepare raw data for analysis. Core analytics methodologies such as classification, clustering, regression, and anomaly detection are examined, with a focus on algorithmic innovation and scalability. Furthermore, the text delves into state-of-the-art frameworks for data mining and predictive modeling, highlighting the role of neural networks, support vector machines, and ensemble methods in tackling complex analytical challenges. Special emphasis is placed on the convergence of big data with distributed computing paradigms, including cloud and edge computing, to address challenges in storage, computation, and real-time analytics. The integration of ethical considerations, including data privacy and compliance with global standards, ensures a holistic perspective on data management. Practical applications across healthcare, finance, marketing, and policy-making illustrate the real-world impact of these technologies. Through comprehensive case studies and Python-based implementations, this work equips researchers, practitioners, and data enthusiasts with the tools to navigate the complexities of modern data analytics. It bridges the gap between theory and practice, fostering the development of innovative solutions for managing and leveraging data in the era of artificial intelligence.
A Comprehensive Guide to Explainable AI: From Classical Models to LLMs
Dec 01, 2024



Explainable Artificial Intelligence (XAI) addresses the growing need for transparency and interpretability in AI systems, enabling trust and accountability in decision-making processes. This book offers a comprehensive guide to XAI, bridging foundational concepts with advanced methodologies. It explores interpretability in traditional models such as Decision Trees, Linear Regression, and Support Vector Machines, alongside the challenges of explaining deep learning architectures like CNNs, RNNs, and Large Language Models (LLMs), including BERT, GPT, and T5. The book presents practical techniques such as SHAP, LIME, Grad-CAM, counterfactual explanations, and causal inference, supported by Python code examples for real-world applications. Case studies illustrate XAI's role in healthcare, finance, and policymaking, demonstrating its impact on fairness and decision support. The book also covers evaluation metrics for explanation quality, an overview of cutting-edge XAI tools and frameworks, and emerging research directions, such as interpretability in federated learning and ethical AI considerations. Designed for a broad audience, this resource equips readers with the theoretical insights and practical skills needed to master XAI. Hands-on examples and additional resources are available at the companion GitHub repository: https://github.com/Echoslayer/XAI_From_Classical_Models_to_LLMs.
Generative Modeling of Molecular Dynamics Trajectories
Sep 26, 2024



Molecular dynamics (MD) is a powerful technique for studying microscopic phenomena, but its computational cost has driven significant interest in the development of deep learning-based surrogate models. We introduce generative modeling of molecular trajectories as a paradigm for learning flexible multi-task surrogate models of MD from data. By conditioning on appropriately chosen frames of the trajectory, we show such generative models can be adapted to diverse tasks such as forward simulation, transition path sampling, and trajectory upsampling. By alternatively conditioning on part of the molecular system and inpainting the rest, we also demonstrate the first steps towards dynamics-conditioned molecular design. We validate the full set of these capabilities on tetrapeptide simulations and show that our model can produce reasonable ensembles of protein monomers. Altogether, our work illustrates how generative modeling can unlock value from MD data towards diverse downstream tasks that are not straightforward to address with existing methods or even MD itself. Code is available at https://github.com/bjing2016/mdgen.
Verlet Flows: Exact-Likelihood Integrators for Flow-Based Generative Models
May 05, 2024


Approximations in computing model likelihoods with continuous normalizing flows (CNFs) hinder the use of these models for importance sampling of Boltzmann distributions, where exact likelihoods are required. In this work, we present Verlet flows, a class of CNFs on an augmented state-space inspired by symplectic integrators from Hamiltonian dynamics. When used with carefully constructed Taylor-Verlet integrators, Verlet flows provide exact-likelihood generative models which generalize coupled flow architectures from a non-continuous setting while imposing minimal expressivity constraints. On experiments over toy densities, we demonstrate that the variance of the commonly used Hutchinson trace estimator is unsuitable for importance sampling, whereas Verlet flows perform comparably to full autograd trace computations while being significantly faster.
Dirichlet Flow Matching with Applications to DNA Sequence Design
Feb 08, 2024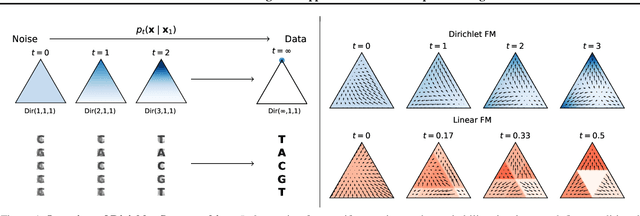

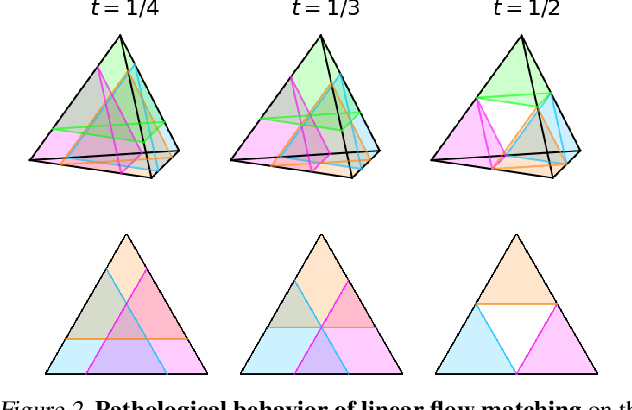
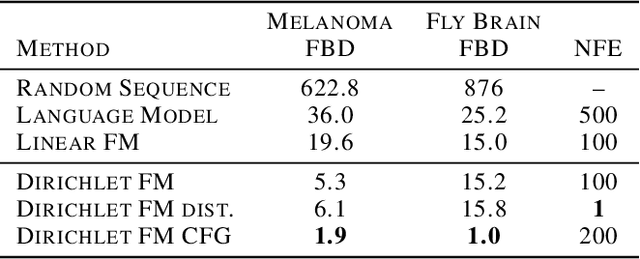
Discrete diffusion or flow models could enable faster and more controllable sequence generation than autoregressive models. We show that na\"ive linear flow matching on the simplex is insufficient toward this goal since it suffers from discontinuities in the training target and further pathologies. To overcome this, we develop Dirichlet flow matching on the simplex based on mixtures of Dirichlet distributions as probability paths. In this framework, we derive a connection between the mixtures' scores and the flow's vector field that allows for classifier and classifier-free guidance. Further, we provide distilled Dirichlet flow matching, which enables one-step sequence generation with minimal performance hits, resulting in $O(L)$ speedups compared to autoregressive models. On complex DNA sequence generation tasks, we demonstrate superior performance compared to all baselines in distributional metrics and in achieving desired design targets for generated sequences. Finally, we show that our classifier-free guidance approach improves unconditional generation and is effective for generating DNA that satisfies design targets. Code is available at https://github.com/HannesStark/dirichlet-flow-matching.
AlphaFold Meets Flow Matching for Generating Protein Ensembles
Feb 07, 2024



The biological functions of proteins often depend on dynamic structural ensembles. In this work, we develop a flow-based generative modeling approach for learning and sampling the conformational landscapes of proteins. We repurpose highly accurate single-state predictors such as AlphaFold and ESMFold and fine-tune them under a custom flow matching framework to obtain sequence-conditoned generative models of protein structure called AlphaFlow and ESMFlow. When trained and evaluated on the PDB, our method provides a superior combination of precision and diversity compared to AlphaFold with MSA subsampling. When further trained on ensembles from all-atom MD, our method accurately captures conformational flexibility, positional distributions, and higher-order ensemble observables for unseen proteins. Moreover, our method can diversify a static PDB structure with faster wall-clock convergence to certain equilibrium properties than replicate MD trajectories, demonstrating its potential as a proxy for expensive physics-based simulations. Code is available at https://github.com/bjing2016/alphaflow.
Equivariant Scalar Fields for Molecular Docking with Fast Fourier Transforms
Dec 07, 2023Molecular docking is critical to structure-based virtual screening, yet the throughput of such workflows is limited by the expensive optimization of scoring functions involved in most docking algorithms. We explore how machine learning can accelerate this process by learning a scoring function with a functional form that allows for more rapid optimization. Specifically, we define the scoring function to be the cross-correlation of multi-channel ligand and protein scalar fields parameterized by equivariant graph neural networks, enabling rapid optimization over rigid-body degrees of freedom with fast Fourier transforms. The runtime of our approach can be amortized at several levels of abstraction, and is particularly favorable for virtual screening settings with a common binding pocket. We benchmark our scoring functions on two simplified docking-related tasks: decoy pose scoring and rigid conformer docking. Our method attains similar but faster performance on crystal structures compared to the widely-used Vina and Gnina scoring functions, and is more robust on computationally predicted structures. Code is available at https://github.com/bjing2016/scalar-fields.
Harmonic Self-Conditioned Flow Matching for Multi-Ligand Docking and Binding Site Design
Oct 09, 2023



A significant amount of protein function requires binding small molecules, including enzymatic catalysis. As such, designing binding pockets for small molecules has several impactful applications ranging from drug synthesis to energy storage. Towards this goal, we first develop HarmonicFlow, an improved generative process over 3D protein-ligand binding structures based on our self-conditioned flow matching objective. FlowSite extends this flow model to jointly generate a protein pocket's discrete residue types and the molecule's binding 3D structure. We show that HarmonicFlow improves upon the state-of-the-art generative processes for docking in simplicity, generality, and performance. Enabled by this structure modeling, FlowSite designs binding sites substantially better than baseline approaches and provides the first general solution for binding site design.