 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePoET: A generative model of protein families as sequences-of-sequences
Jun 09, 2023



Generative protein language models are a natural way to design new proteins with desired functions. However, current models are either difficult to direct to produce a protein from a specific family of interest, or must be trained on a large multiple sequence alignment (MSA) from the specific family of interest, making them unable to benefit from transfer learning across families. To address this, we propose $\textbf{P}$r$\textbf{o}$tein $\textbf{E}$volutionary $\textbf{T}$ransformer (PoET), an autoregressive generative model of whole protein families that learns to generate sets of related proteins as sequences-of-sequences across tens of millions of natural protein sequence clusters. PoET can be used as a retrieval-augmented language model to generate and score arbitrary modifications conditioned on any protein family of interest, and can extrapolate from short context lengths to generalize well even for small families. This is enabled by a unique Transformer layer; we model tokens sequentially within sequences while attending between sequences order invariantly, allowing PoET to scale to context lengths beyond those used during training. PoET outperforms existing protein language models and evolutionary sequence models for variant function prediction in extensive experiments on deep mutational scanning datasets, improving variant effect prediction across proteins of all MSA depths.
Unsupervised Object Representation Learning using Translation and Rotation Group Equivariant VAE
Oct 24, 2022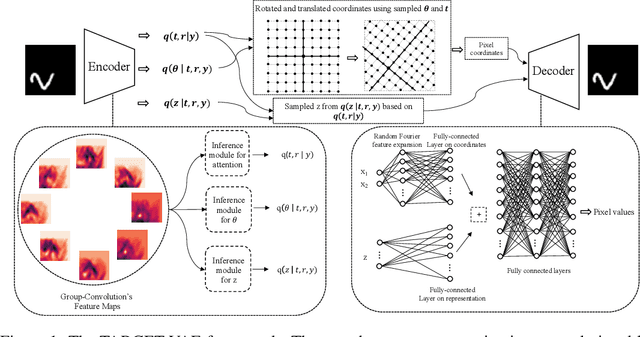


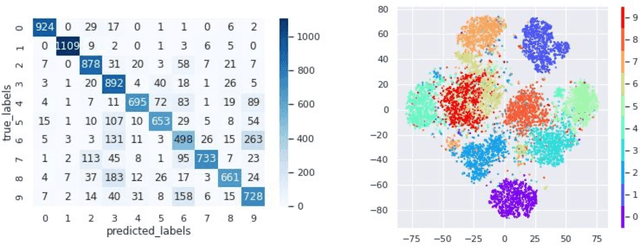
In many imaging modalities, objects of interest can occur in a variety of locations and poses (i.e. are subject to translations and rotations in 2d or 3d), but the location and pose of an object does not change its semantics (i.e. the object's essence). That is, the specific location and rotation of an airplane in satellite imagery, or the 3d rotation of a chair in a natural image, or the rotation of a particle in a cryo-electron micrograph, do not change the intrinsic nature of those objects. Here, we consider the problem of learning semantic representations of objects that are invariant to pose and location in a fully unsupervised manner. We address shortcomings in previous approaches to this problem by introducing TARGET-VAE, a translation and rotation group-equivariant variational autoencoder framework. TARGET-VAE combines three core innovations: 1) a rotation and translation group-equivariant encoder architecture, 2) a structurally disentangled distribution over latent rotation, translation, and a rotation-translation-invariant semantic object representation, which are jointly inferred by the approximate inference network, and 3) a spatially equivariant generator network. In comprehensive experiments, we show that TARGET-VAE learns disentangled representations without supervision that significantly improve upon, and avoid the pathologies of, previous methods. When trained on images highly corrupted by rotation and translation, the semantic representations learned by TARGET-VAE are similar to those learned on consistently posed objects, dramatically improving clustering in the semantic latent space. Furthermore, TARGET-VAE is able to perform remarkably accurate unsupervised pose and location inference. We expect methods like TARGET-VAE will underpin future approaches for unsupervised object generation, pose prediction, and object detection.
Antibody Representation Learning for Drug Discovery
Oct 05, 2022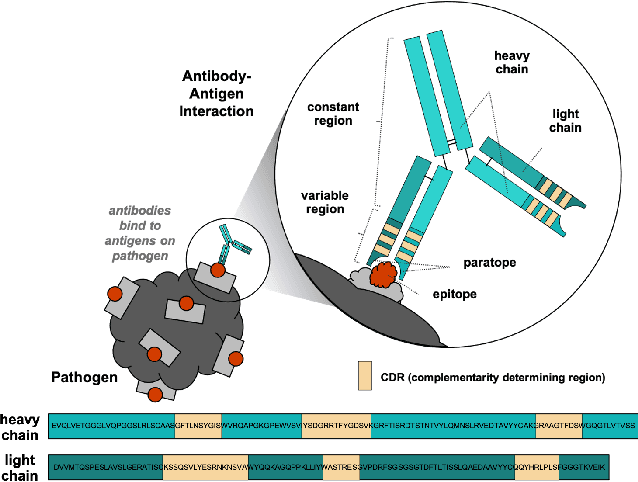
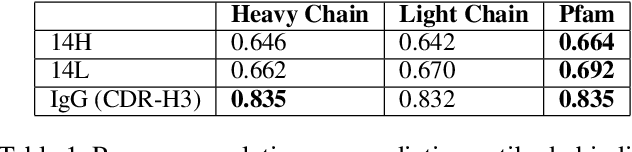
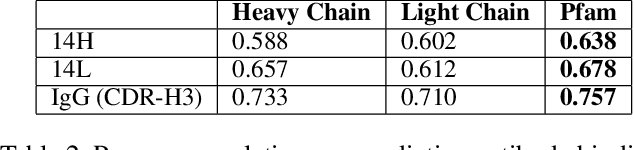
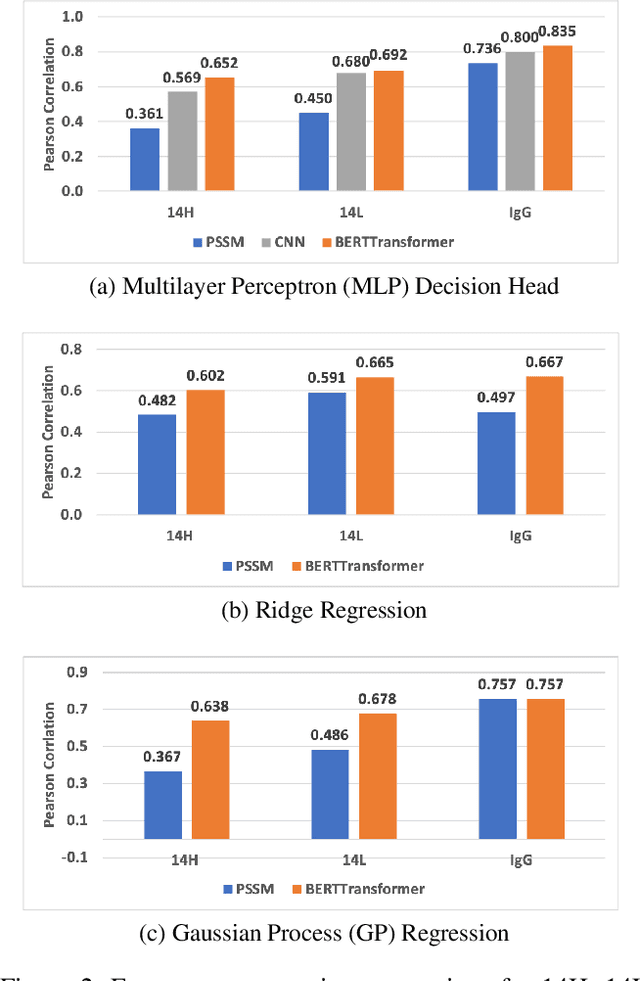
Therapeutic antibody development has become an increasingly popular approach for drug development. To date, antibody therapeutics are largely developed using large scale experimental screens of antibody libraries containing hundreds of millions of antibody sequences. The high cost and difficulty of developing therapeutic antibodies create a pressing need for computational methods to predict antibody properties and create bespoke designs. However, the relationship between antibody sequence and activity is a complex physical process and traditional iterative design approaches rely on large scale assays and random mutagenesis. Deep learning methods have emerged as a promising way to learn antibody property predictors, but predicting antibody properties and target-specific activities depends critically on the choice of antibody representations and data linking sequences to properties is often limited. Existing works have not yet investigated the value, limitations and opportunities of these methods in application to antibody-based drug discovery. In this paper, we present results on a novel SARS-CoV-2 antibody binding dataset and an additional benchmark dataset. We compare three classes of models: conventional statistical sequence models, supervised learning on each dataset independently, and fine-tuning an antibody specific pre-trained language model. Experimental results suggest that self-supervised pretraining of feature representation consistently offers significant improvement in over previous approaches. We also investigate the impact of data size on the model performance, and discuss challenges and opportunities that the machine learning community can address to advance in silico engineering and design of therapeutic antibodies.
Few Shot Protein Generation
Apr 03, 2022



We present the MSA-to-protein transformer, a generative model of protein sequences conditioned on protein families represented by multiple sequence alignments (MSAs). Unlike existing approaches to learning generative models of protein families, the MSA-to-protein transformer conditions sequence generation directly on a learned encoding of the multiple sequence alignment, circumventing the need for fitting dedicated family models. By training on a large set of well-curated multiple sequence alignments in Pfam, our MSA-to-protein transformer generalizes well to protein families not observed during training and outperforms conventional family modeling approaches, especially when MSAs are small. Our generative approach accurately models epistasis and indels and allows for exact inference and efficient sampling unlike other approaches. We demonstrate the protein sequence modeling capabilities of our MSA-to-protein transformer and compare it with alternative sequence modeling approaches in comprehensive benchmark experiments.
Learning to automate cryo-electron microscopy data collection with Ptolemy
Dec 01, 2021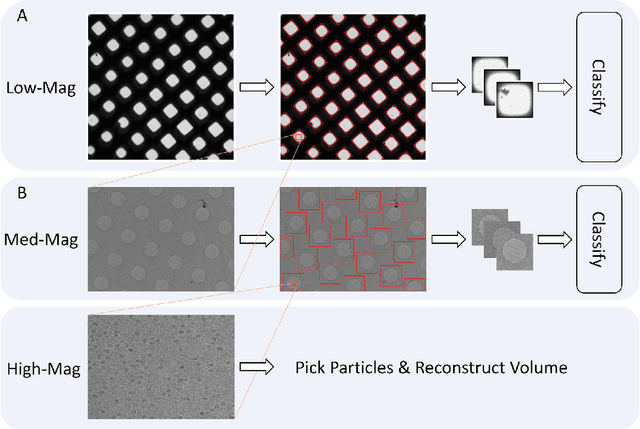


Over the past decade, cryogenic electron microscopy (cryo-EM) has emerged as a primary method for determining near-native, near-atomic resolution 3D structures of biological macromolecules. In order to meet increasing demand for cryo-EM, automated methods to improve throughput and efficiency while lowering costs are needed. Currently, the process of collecting high-magnification cryo-EM micrographs, data collection, requires human input and manual tuning of parameters, as expert operators must navigate low- and medium-magnification images to find good high-magnification collection locations. Automating this is non-trivial: the images suffer from low signal-to-noise ratio and are affected by a range of experimental parameters that can differ for each collection session. Here, we use various computer vision algorithms, including mixture models, convolutional neural networks (CNNs), and U-Nets to develop the first pipeline to automate low- and medium-magnification targeting with purpose-built algorithms. Learned models in this pipeline are trained on a large internal dataset of images from real world cryo-EM data collection sessions, labeled with locations that were selected by operators. Using these models, we show that we can effectively detect and classify regions of interest (ROIs) in low- and medium-magnification images, and can generalize to unseen sessions, as well as to images captured using different microscopes from external facilities. We expect our pipeline, Ptolemy, will be both immediately useful as a tool for automation of cryo-EM data collection, and serve as a foundation for future advanced methods for efficient and automated cryo-EM microscopy.
Explicitly disentangling image content from translation and rotation with spatial-VAE
Sep 25, 2019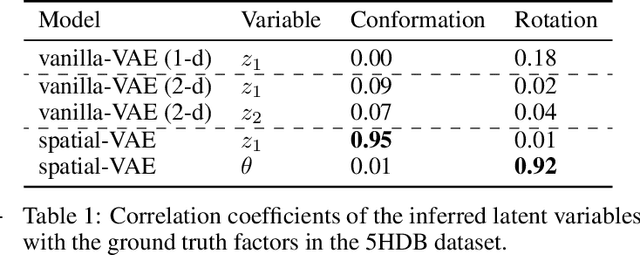
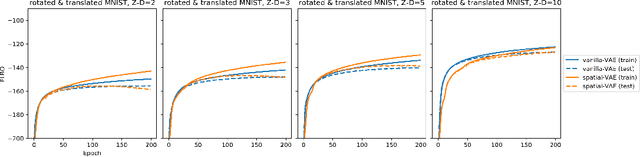

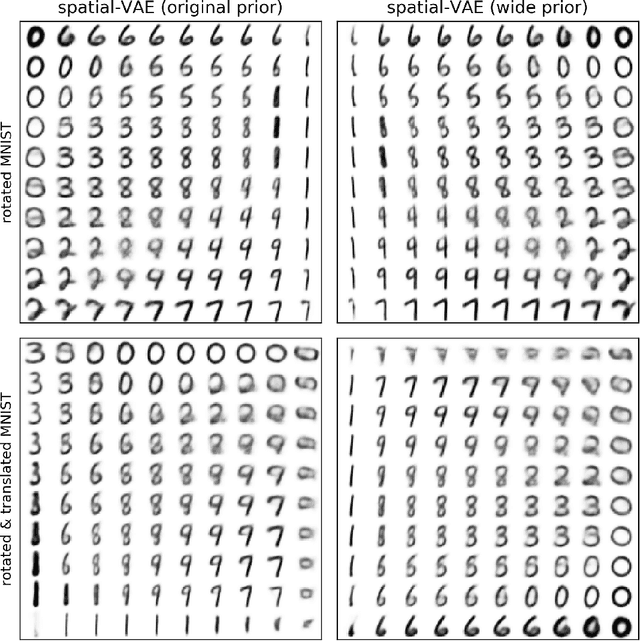
Given an image dataset, we are often interested in finding data generative factors that encode semantic content independently from pose variables such as rotation and translation. However, current disentanglement approaches do not impose any specific structure on the learned latent representations. We propose a method for explicitly disentangling image rotation and translation from other unstructured latent factors in a variational autoencoder (VAE) framework. By formulating the generative model as a function of the spatial coordinate, we make the reconstruction error differentiable with respect to latent translation and rotation parameters. This formulation allows us to train a neural network to perform approximate inference on these latent variables while explicitly constraining them to only represent rotation and translation. We demonstrate that this framework, termed spatial-VAE, effectively learns latent representations that disentangle image rotation and translation from content and improves reconstruction over standard VAEs on several benchmark datasets, including applications to modeling continuous 2-D views of proteins from single particle electron microscopy and galaxies in astronomical images.
Reconstructing continuously heterogeneous structures from single particle cryo-EM with deep generative models
Sep 11, 2019



Cryo-electron microscopy (cryo-EM) is a powerful technique for determining the structure of proteins and other macromolecular complexes at near-atomic resolution. In single particle cryo-EM, the central problem is to reconstruct the three-dimensional structure of a macromolecule from $10^{4-7}$ noisy and randomly oriented two-dimensional projections. However, the imaged protein complexes may exhibit structural variability, which complicates reconstruction and is typically addressed using discrete clustering approaches that fail to capture the full range of protein dynamics. Here, we introduce a novel method for cryo-EM reconstruction that extends naturally to modeling continuous generative factors of structural heterogeneity. This method encodes structures in Fourier space using coordinate-based deep neural networks, and trains these networks from unlabeled 2D cryo-EM images by combining exact inference over image orientation with variational inference for structural heterogeneity. We demonstrate that the proposed method, termed cryoDRGN, can perform ab initio reconstruction of 3D protein complexes from simulated and real 2D cryo-EM image data. To our knowledge, cryoDRGN is the first neural network-based approach for cryo-EM reconstruction and the first end-to-end method for directly reconstructing continuous ensembles of protein structures from cryo-EM images.
Learning protein sequence embeddings using information from structure
Feb 22, 2019


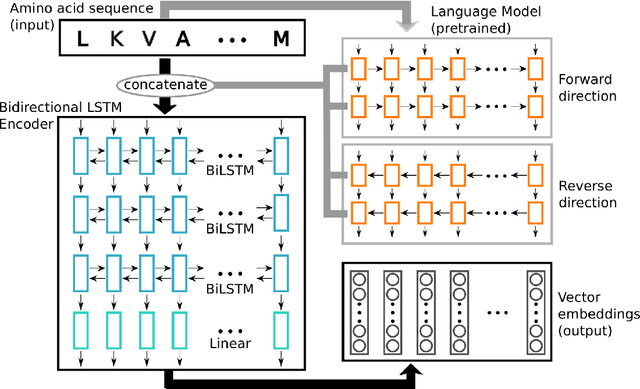
Inferring the structural properties of a protein from its amino acid sequence is a challenging yet important problem in biology. Structures are not known for the vast majority of protein sequences, but structure is critical for understanding function. Existing approaches for detecting structural similarity between proteins from sequence are unable to recognize and exploit structural patterns when sequences have diverged too far, limiting our ability to transfer knowledge between structurally related proteins. We newly approach this problem through the lens of representation learning. We introduce a framework that maps any protein sequence to a sequence of vector embeddings --- one per amino acid position --- that encode structural information. We train bidirectional long short-term memory (LSTM) models on protein sequences with a two-part feedback mechanism that incorporates information from (i) global structural similarity between proteins and (ii) pairwise residue contact maps for individual proteins. To enable learning from structural similarity information, we define a novel similarity measure between arbitrary-length sequences of vector embeddings based on a soft symmetric alignment (SSA) between them. Our method is able to learn useful position-specific embeddings despite lacking direct observations of position-level correspondence between sequences. We show empirically that our multi-task framework outperforms other sequence-based methods and even a top-performing structure-based alignment method when predicting structural similarity, our goal. Finally, we demonstrate that our learned embeddings can be transferred to other protein sequence problems, improving the state-of-the-art in transmembrane domain prediction.
* 17 pages, 3 figures, 8 tables, proceedings of ICLR 2019
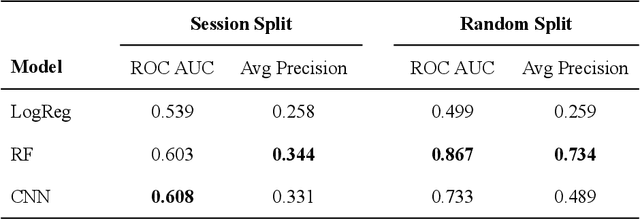
Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs
Oct 08, 2018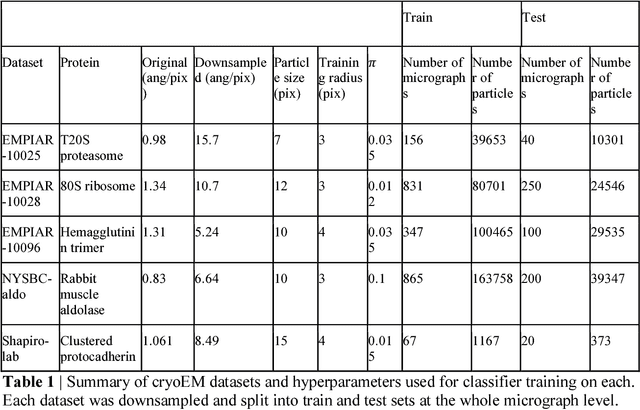
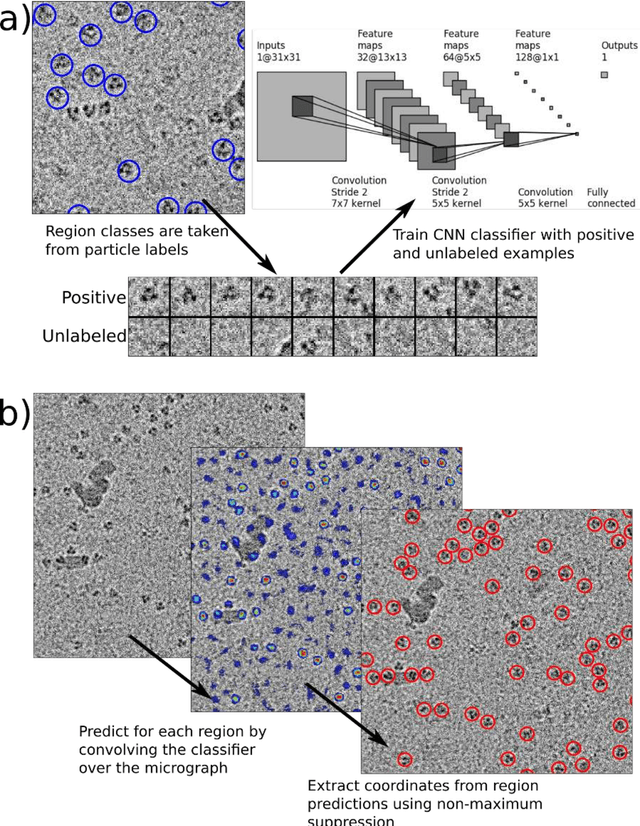
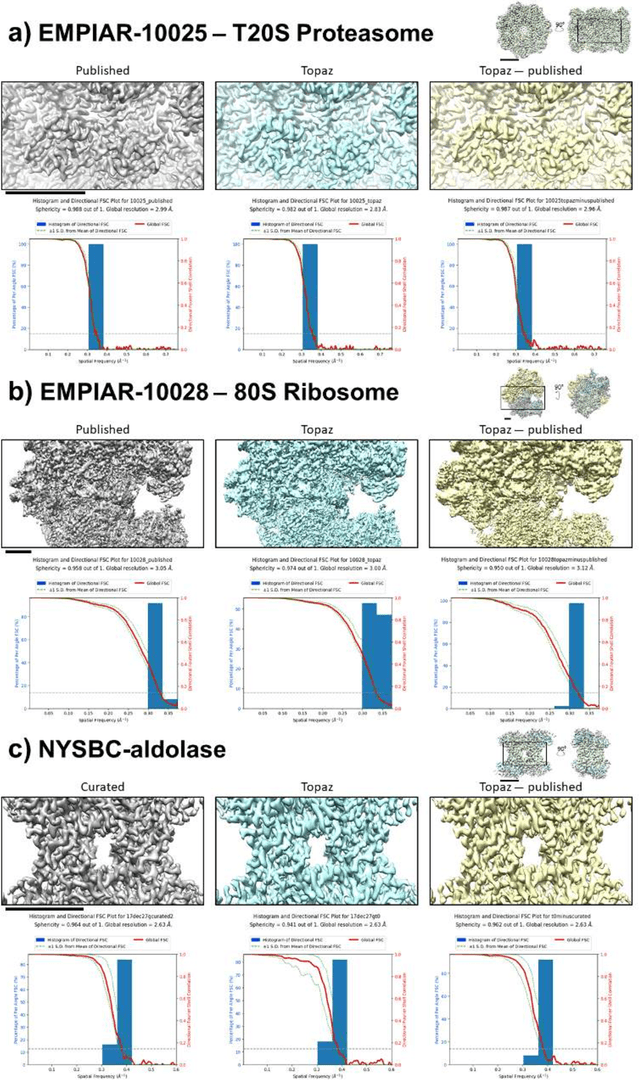
Cryo-electron microscopy (cryoEM) is an increasingly popular method for protein structure determination. However, identifying a sufficient number of particles for analysis (often >100,000) can take months of manual effort. Current computational approaches are limited by high false positive rates and require significant ad-hoc post-processing, especially for unusually shaped particles. To address this shortcoming, we develop Topaz, an efficient and accurate particle picking pipeline using neural networks trained with few labeled particles by newly leveraging the remaining unlabeled particles through the framework of positive-unlabeled (PU) learning. Remarkably, despite using minimal labeled particles, Topaz allows us to improve reconstruction resolution by up to 0.15 {\AA} over published particles on three public cryoEM datasets without any post-processing. Furthermore, we show that our novel generalized-expectation criteria approach to PU learning outperforms existing general PU learning approaches when applied to particle detection, especially for challenging datasets of non-globular proteins. We expect Topaz to be an essential component of cryoEM analysis.