 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSTEP3-VL-10B Technical Report
Jan 15, 2026We present STEP3-VL-10B, a lightweight open-source foundation model designed to redefine the trade-off between compact efficiency and frontier-level multimodal intelligence. STEP3-VL-10B is realized through two strategic shifts: first, a unified, fully unfrozen pre-training strategy on 1.2T multimodal tokens that integrates a language-aligned Perception Encoder with a Qwen3-8B decoder to establish intrinsic vision-language synergy; and second, a scaled post-training pipeline featuring over 1k iterations of reinforcement learning. Crucially, we implement Parallel Coordinated Reasoning (PaCoRe) to scale test-time compute, allocating resources to scalable perceptual reasoning that explores and synthesizes diverse visual hypotheses. Consequently, despite its compact 10B footprint, STEP3-VL-10B rivals or surpasses models 10$\times$-20$\times$ larger (e.g., GLM-4.6V-106B, Qwen3-VL-235B) and top-tier proprietary flagships like Gemini 2.5 Pro and Seed-1.5-VL. Delivering best-in-class performance, it records 92.2% on MMBench and 80.11% on MMMU, while excelling in complex reasoning with 94.43% on AIME2025 and 75.95% on MathVision. We release the full model suite to provide the community with a powerful, efficient, and reproducible baseline.
Evolutionary Profiles for Protein Fitness Prediction
Oct 08, 2025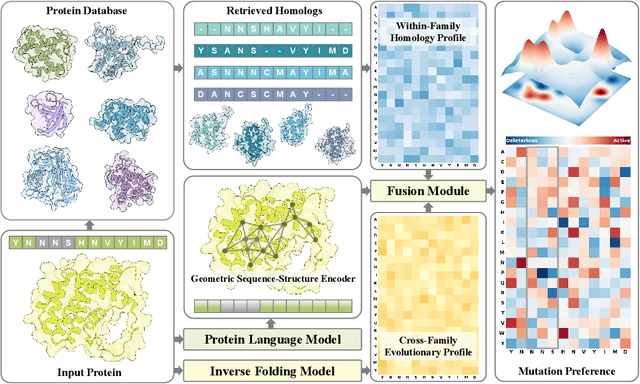
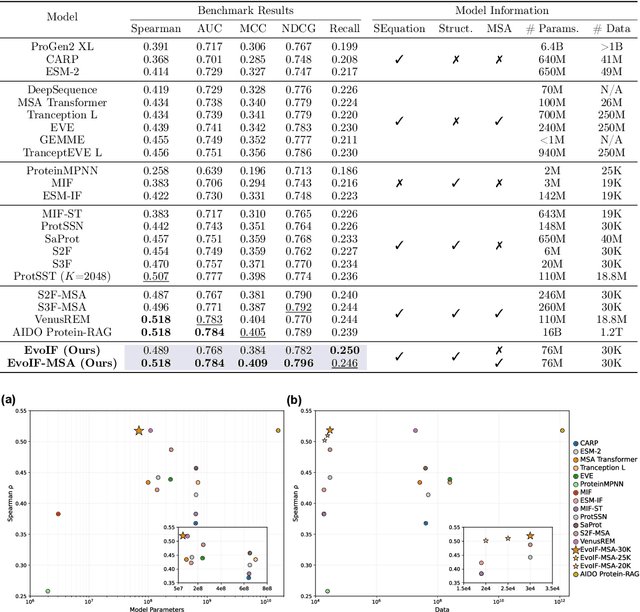
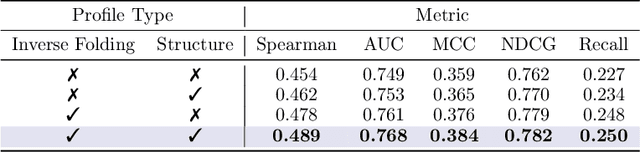
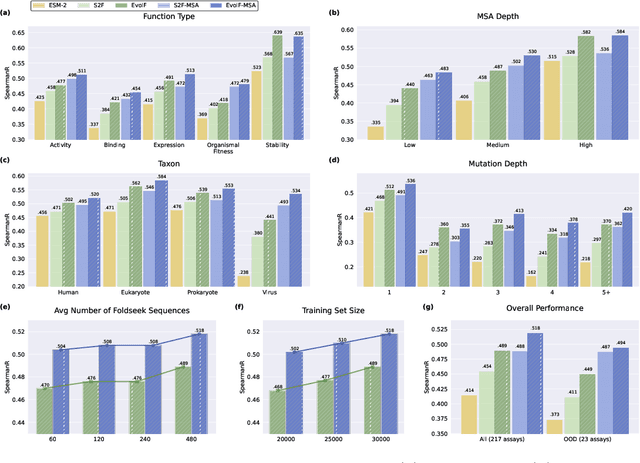
Predicting the fitness impact of mutations is central to protein engineering but constrained by limited assays relative to the size of sequence space. Protein language models (pLMs) trained with masked language modeling (MLM) exhibit strong zero-shot fitness prediction; we provide a unifying view by interpreting natural evolution as implicit reward maximization and MLM as inverse reinforcement learning (IRL), in which extant sequences act as expert demonstrations and pLM log-odds serve as fitness estimates. Building on this perspective, we introduce EvoIF, a lightweight model that integrates two complementary sources of evolutionary signal: (i) within-family profiles from retrieved homologs and (ii) cross-family structural-evolutionary constraints distilled from inverse folding logits. EvoIF fuses sequence-structure representations with these profiles via a compact transition block, yielding calibrated probabilities for log-odds scoring. On ProteinGym (217 mutational assays; >2.5M mutants), EvoIF and its MSA-enabled variant achieve state-of-the-art or competitive performance while using only 0.15% of the training data and fewer parameters than recent large models. Ablations confirm that within-family and cross-family profiles are complementary, improving robustness across function types, MSA depths, taxa, and mutation depths. The codes will be made publicly available at https://github.com/aim-uofa/EvoIF.
Boltzmann-Aligned Inverse Folding Model as a Predictor of Mutational Effects on Protein-Protein Interactions
Oct 12, 2024



Predicting the change in binding free energy ($\Delta \Delta G$) is crucial for understanding and modulating protein-protein interactions, which are critical in drug design. Due to the scarcity of experimental $\Delta \Delta G$ data, existing methods focus on pre-training, while neglecting the importance of alignment. In this work, we propose the Boltzmann Alignment technique to transfer knowledge from pre-trained inverse folding models to $\Delta \Delta G$ prediction. We begin by analyzing the thermodynamic definition of $\Delta \Delta G$ and introducing the Boltzmann distribution to connect energy with protein conformational distribution. However, the protein conformational distribution is intractable; therefore, we employ Bayes' theorem to circumvent direct estimation and instead utilize the log-likelihood provided by protein inverse folding models for $\Delta \Delta G$ estimation. Compared to previous inverse folding-based methods, our method explicitly accounts for the unbound state of protein complex in the $\Delta \Delta G$ thermodynamic cycle, introducing a physical inductive bias and achieving both supervised and unsupervised state-of-the-art (SoTA) performance. Experimental results on SKEMPI v2 indicate that our method achieves Spearman coefficients of 0.3201 (unsupervised) and 0.5134 (supervised), significantly surpassing the previously reported SoTA values of 0.2632 and 0.4324, respectively. Futhermore, we demonstrate the capability of our method on binding energy prediction, protein-protein docking and antibody optimization tasks.
Floating Anchor Diffusion Model for Multi-motif Scaffolding
Jun 05, 2024



Motif scaffolding seeks to design scaffold structures for constructing proteins with functions derived from the desired motif, which is crucial for the design of vaccines and enzymes. Previous works approach the problem by inpainting or conditional generation. Both of them can only scaffold motifs with fixed positions, and the conditional generation cannot guarantee the presence of motifs. However, prior knowledge of the relative motif positions in a protein is not readily available, and constructing a protein with multiple functions in one protein is more general and significant because of the synergies between functions. We propose a Floating Anchor Diffusion (FADiff) model. FADiff allows motifs to float rigidly and independently in the process of diffusion, which guarantees the presence of motifs and automates the motif position design. Our experiments demonstrate the efficacy of FADiff with high success rates and designable novel scaffolds. To the best of our knowledge, FADiff is the first work to tackle the challenge of scaffolding multiple motifs without relying on the expertise of relative motif positions in the protein. Code is available at https://github.com/aim-uofa/FADiff.