 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDPA-1: Pretraining of Attention-based Deep Potential Model for Molecular Simulation
Aug 20, 2022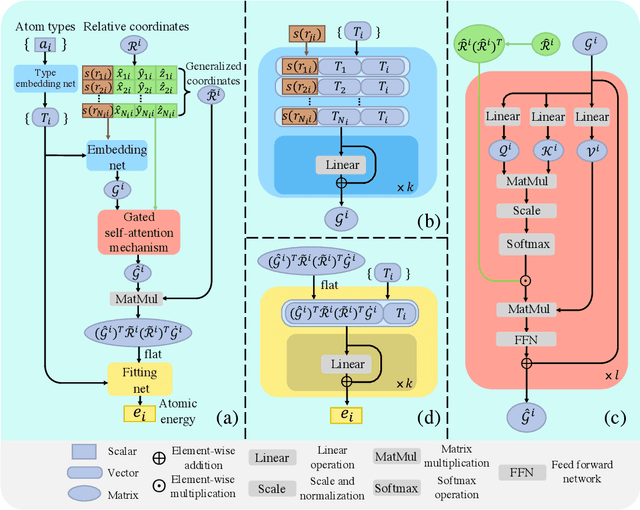
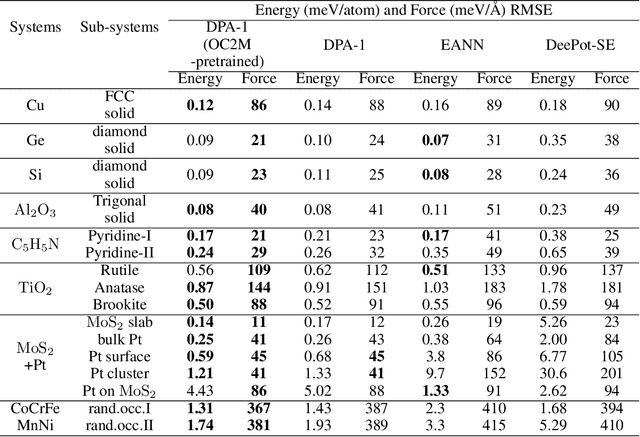
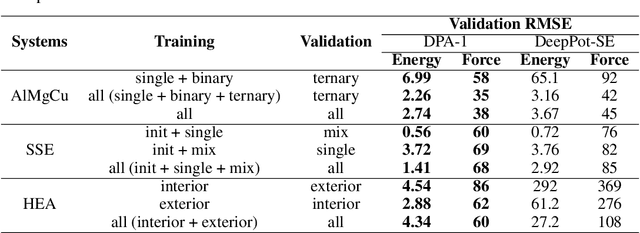
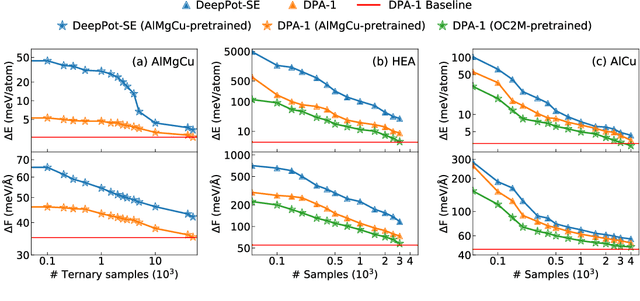
Machine learning assisted modeling of the inter-atomic potential energy surface (PES) is revolutionizing the field of molecular simulation. With the accumulation of high-quality electronic structure data, a model that can be pretrained on all available data and finetuned on downstream tasks with a small additional effort would bring the field to a new stage. Here we propose DPA-1, a Deep Potential model with a novel attention mechanism, which is highly effective for representing the conformation and chemical spaces of atomic systems and learning the PES. We tested DPA-1 on a number of systems and observed superior performance compared with existing benchmarks. When pretrained on large-scale datasets containing 56 elements, DPA-1 can be successfully applied to various downstream tasks with a great improvement of sample efficiency. Surprisingly, for different elements, the learned type embedding parameters form a $spiral$ in the latent space and have a natural correspondence with their positions on the periodic table, showing interesting interpretability of the pretrained DPA-1 model.
Non-Autoregressive Electron Redistribution Modeling for Reaction Prediction
Jun 08, 2021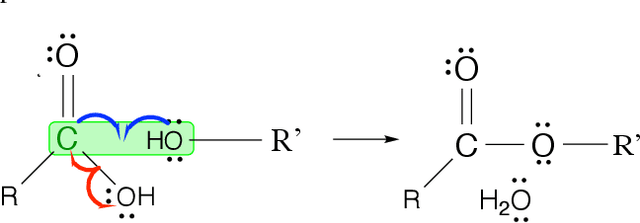
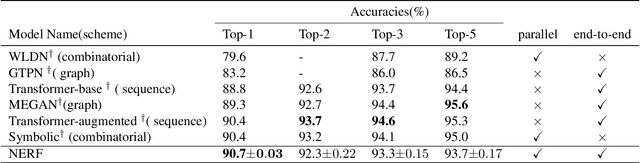
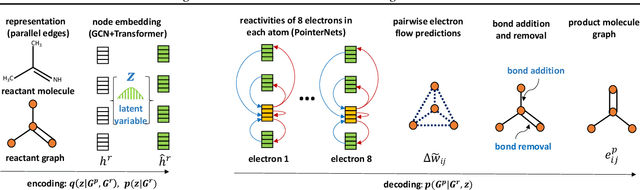
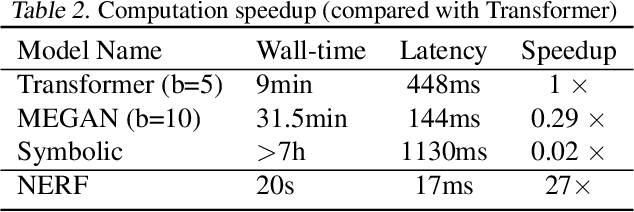
Reliably predicting the products of chemical reactions presents a fundamental challenge in synthetic chemistry. Existing machine learning approaches typically produce a reaction product by sequentially forming its subparts or intermediate molecules. Such autoregressive methods, however, not only require a pre-defined order for the incremental construction but preclude the use of parallel decoding for efficient computation. To address these issues, we devise a non-autoregressive learning paradigm that predicts reaction in one shot. Leveraging the fact that chemical reactions can be described as a redistribution of electrons in molecules, we formulate a reaction as an arbitrary electron flow and predict it with a novel multi-pointer decoding network. Experiments on the USPTO-MIT dataset show that our approach has established a new state-of-the-art top-1 accuracy and achieves at least 27 times inference speedup over the state-of-the-art methods. Also, our predictions are easier for chemists to interpret owing to predicting the electron flows.
Non-autoregressive electron flow generation for reaction prediction
Dec 16, 2020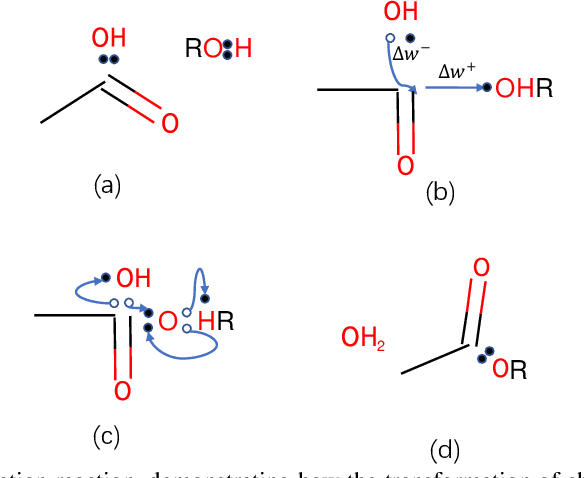
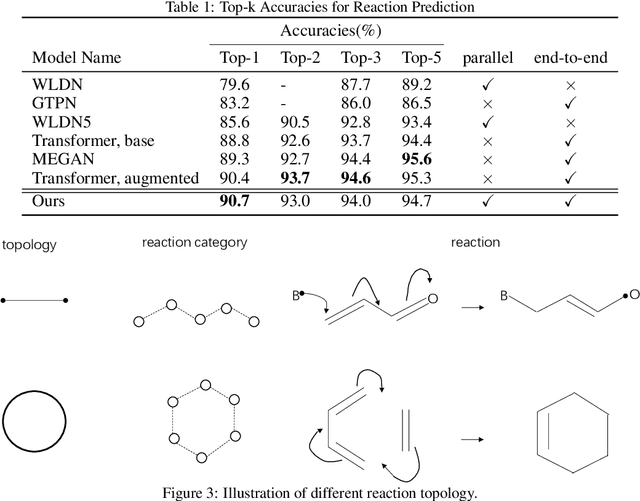
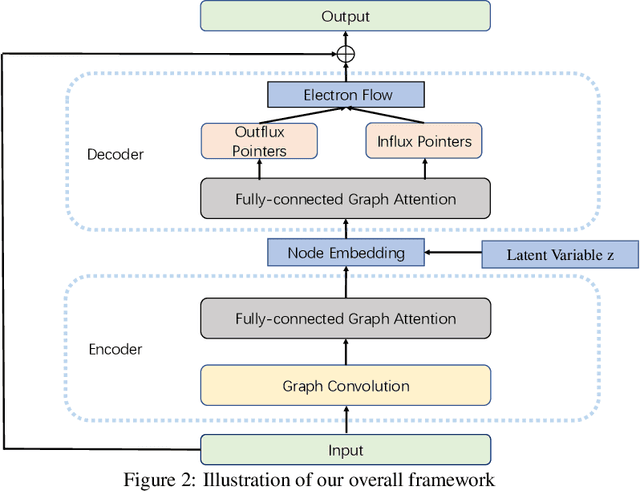

Reaction prediction is a fundamental problem in computational chemistry. Existing approaches typically generate a chemical reaction by sampling tokens or graph edits sequentially, conditioning on previously generated outputs. These autoregressive generating methods impose an arbitrary ordering of outputs and prevent parallel decoding during inference. We devise a novel decoder that avoids such sequential generating and predicts the reaction in a Non-Autoregressive manner. Inspired by physical-chemistry insights, we represent edge edits in a molecule graph as electron flows, which can then be predicted in parallel. To capture the uncertainty of reactions, we introduce latent variables to generate multi-modal outputs. Following previous works, we evaluate our model on USPTO MIT dataset. Our model achieves both an order of magnitude lower inference latency, with state-of-the-art top-1 accuracy and comparable performance on Top-K sampling.