 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDPA-1: Pretraining of Attention-based Deep Potential Model for Molecular Simulation
Paper and Code
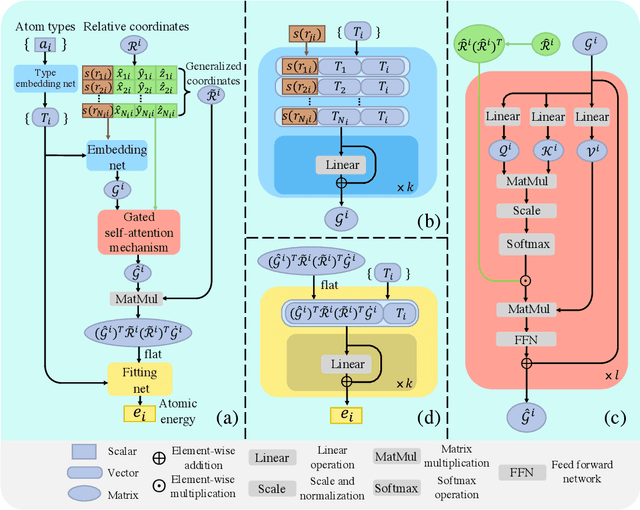
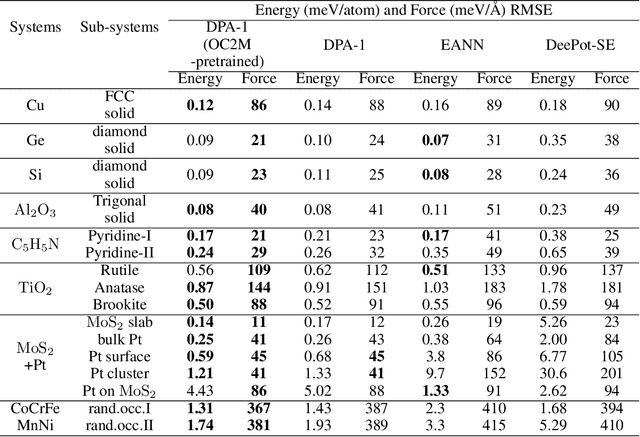
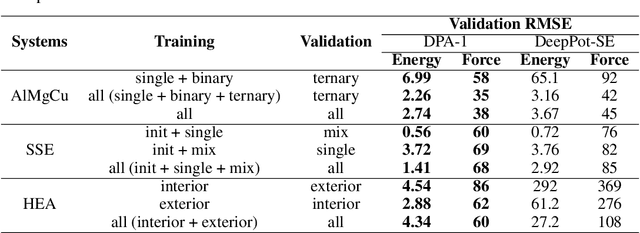
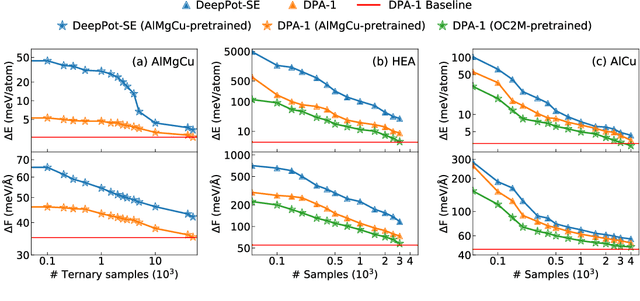
Machine learning assisted modeling of the inter-atomic potential energy surface (PES) is revolutionizing the field of molecular simulation. With the accumulation of high-quality electronic structure data, a model that can be pretrained on all available data and finetuned on downstream tasks with a small additional effort would bring the field to a new stage. Here we propose DPA-1, a Deep Potential model with a novel attention mechanism, which is highly effective for representing the conformation and chemical spaces of atomic systems and learning the PES. We tested DPA-1 on a number of systems and observed superior performance compared with existing benchmarks. When pretrained on large-scale datasets containing 56 elements, DPA-1 can be successfully applied to various downstream tasks with a great improvement of sample efficiency. Surprisingly, for different elements, the learned type embedding parameters form a $spiral$ in the latent space and have a natural correspondence with their positions on the periodic table, showing interesting interpretability of the pretrained DPA-1 model.