 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeE3Bind: An End-to-End Equivariant Network for Protein-Ligand Docking
Paper and Code
Oct 12, 2022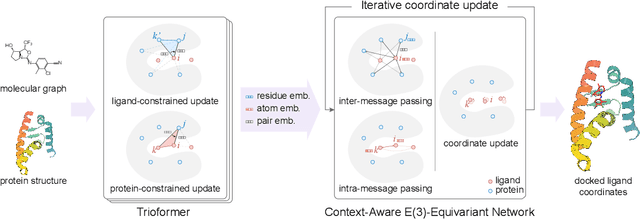
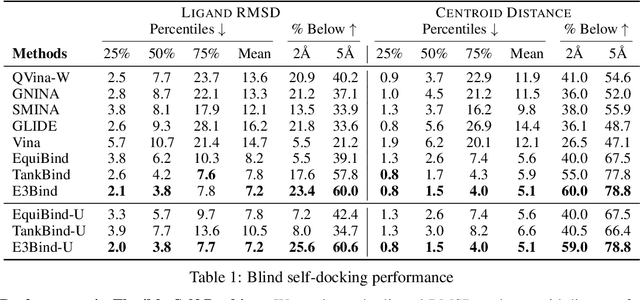
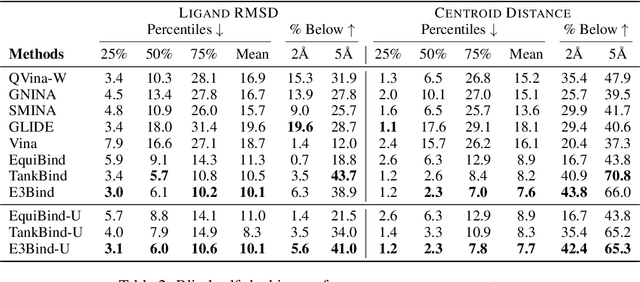
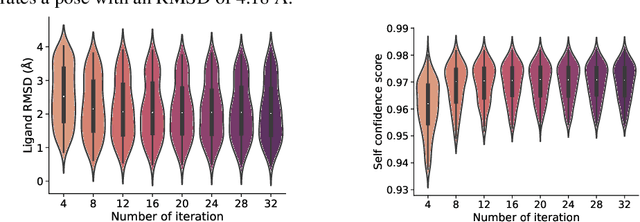
In silico prediction of the ligand binding pose to a given protein target is a crucial but challenging task in drug discovery. This work focuses on blind flexible selfdocking, where we aim to predict the positions, orientations and conformations of docked molecules. Traditional physics-based methods usually suffer from inaccurate scoring functions and high inference cost. Recently, data-driven methods based on deep learning techniques are attracting growing interest thanks to their efficiency during inference and promising performance. These methods usually either adopt a two-stage approach by first predicting the distances between proteins and ligands and then generating the final coordinates based on the predicted distances, or directly predicting the global roto-translation of ligands. In this paper, we take a different route. Inspired by the resounding success of AlphaFold2 for protein structure prediction, we propose E3Bind, an end-to-end equivariant network that iteratively updates the ligand pose. E3Bind models the protein-ligand interaction through careful consideration of the geometric constraints in docking and the local context of the binding site. Experiments on standard benchmark datasets demonstrate the superior performance of our end-to-end trainable model compared to traditional and recently-proposed deep learning methods.