 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeElucidating the Design Space of Multimodal Protein Language Models
Apr 16, 2025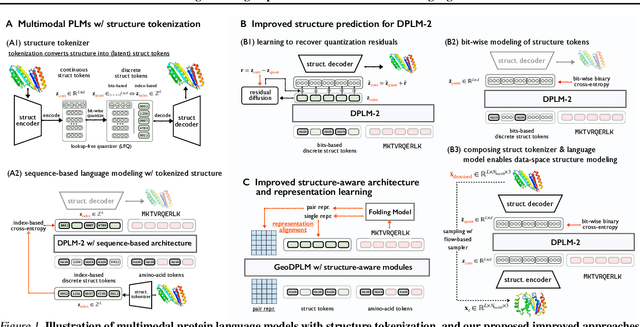
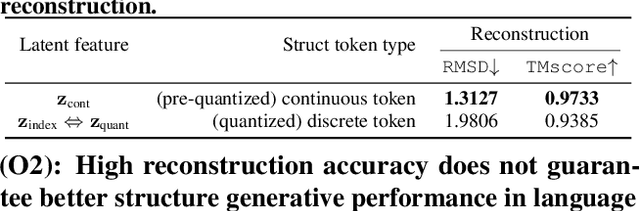
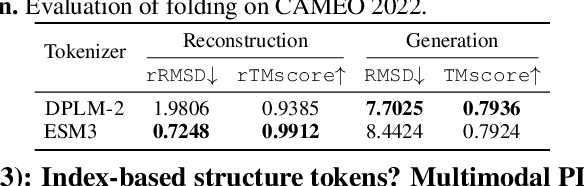
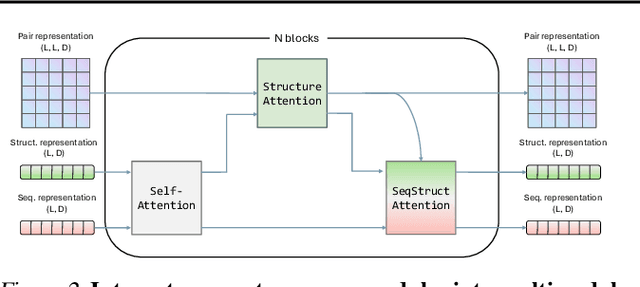
Multimodal protein language models (PLMs) integrate sequence and token-based structural information, serving as a powerful foundation for protein modeling, generation, and design. However, the reliance on tokenizing 3D structures into discrete tokens causes substantial loss of fidelity about fine-grained structural details and correlations. In this paper, we systematically elucidate the design space of multimodal PLMs to overcome their limitations. We identify tokenization loss and inaccurate structure token predictions by the PLMs as major bottlenecks. To address these, our proposed design space covers improved generative modeling, structure-aware architectures and representation learning, and data exploration. Our advancements approach finer-grained supervision, demonstrating that token-based multimodal PLMs can achieve robust structural modeling. The effective design methods dramatically improve the structure generation diversity, and notably, folding abilities of our 650M model by reducing the RMSD from 5.52 to 2.36 on PDB testset, even outperforming 3B baselines and on par with the specialized folding models.
Leveraging Multimodal Protein Representations to Predict Protein Melting Temperatures
Dec 05, 2024



Accurately predicting protein melting temperature changes (Delta Tm) is fundamental for assessing protein stability and guiding protein engineering. Leveraging multi-modal protein representations has shown great promise in capturing the complex relationships among protein sequences, structures, and functions. In this study, we develop models based on powerful protein language models, including ESM-2, ESM-3, SaProt, and AlphaFold, using various feature extraction methods to enhance prediction accuracy. By utilizing the ESM-3 model, we achieve a new state-of-the-art performance on the s571 test dataset, obtaining a Pearson correlation coefficient (PCC) of 0.50. Furthermore, we conduct a fair evaluation to compare the performance of different protein language models in the Delta Tm prediction task. Our results demonstrate that integrating multi-modal protein representations could advance the prediction of protein melting temperatures.
Rectified Flow For Structure Based Drug Design
Dec 02, 2024Deep generative models have achieved tremendous success in structure-based drug design in recent years, especially for generating 3D ligand molecules that bind to specific protein pocket. Notably, diffusion models have transformed ligand generation by providing exceptional quality and creativity. However, traditional diffusion models are restricted by their conventional learning objectives, which limit their broader applicability. In this work, we propose a new framework FlowSBDD, which is based on rectified flow model, allows us to flexibly incorporate additional loss to optimize specific target and introduce additional condition either as an extra input condition or replacing the initial Gaussian distribution. Extensive experiments on CrossDocked2020 show that our approach could achieve state-of-the-art performance on generating high-affinity molecules while maintaining proper molecular properties without specifically designing binding site, with up to -8.50 Avg. Vina Dock score and 75.0% Diversity.