 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSelf-Controlled Dynamic Expansion Model for Continual Learning
Apr 16, 2025Continual Learning (CL) epitomizes an advanced training paradigm wherein prior data samples remain inaccessible during the acquisition of new tasks. Numerous investigations have delved into leveraging a pre-trained Vision Transformer (ViT) to enhance model efficacy in continual learning. Nonetheless, these approaches typically utilize a singular, static backbone, which inadequately adapts to novel tasks, particularly when engaging with diverse data domains, due to a substantial number of inactive parameters. This paper addresses this limitation by introducing an innovative Self-Controlled Dynamic Expansion Model (SCDEM), which orchestrates multiple distinct trainable pre-trained ViT backbones to furnish diverse and semantically enriched representations. Specifically, by employing the multi-backbone architecture as a shared module, the proposed SCDEM dynamically generates a new expert with minimal parameters to accommodate a new task. A novel Collaborative Optimization Mechanism (COM) is introduced to synergistically optimize multiple backbones by harnessing prediction signals from historical experts, thereby facilitating new task learning without erasing previously acquired knowledge. Additionally, a novel Feature Distribution Consistency (FDC) approach is proposed to align semantic similarity between previously and currently learned representations through an optimal transport distance-based mechanism, effectively mitigating negative knowledge transfer effects. Furthermore, to alleviate over-regularization challenges, this paper presents a novel Dynamic Layer-Wise Feature Attention Mechanism (DLWFAM) to autonomously determine the penalization intensity on each trainable representation layer. An extensive series of experiments have been conducted to evaluate the proposed methodology's efficacy, with empirical results corroborating that the approach attains state-of-the-art performance.
Elucidating the Design Space of Multimodal Protein Language Models
Apr 16, 2025Multimodal protein language models (PLMs) integrate sequence and token-based structural information, serving as a powerful foundation for protein modeling, generation, and design. However, the reliance on tokenizing 3D structures into discrete tokens causes substantial loss of fidelity about fine-grained structural details and correlations. In this paper, we systematically elucidate the design space of multimodal PLMs to overcome their limitations. We identify tokenization loss and inaccurate structure token predictions by the PLMs as major bottlenecks. To address these, our proposed design space covers improved generative modeling, structure-aware architectures and representation learning, and data exploration. Our advancements approach finer-grained supervision, demonstrating that token-based multimodal PLMs can achieve robust structural modeling. The effective design methods dramatically improve the structure generation diversity, and notably, folding abilities of our 650M model by reducing the RMSD from 5.52 to 2.36 on PDB testset, even outperforming 3B baselines and on par with the specialized folding models.
Bayesian Neural Networks for One-to-Many Mapping in Image Enhancement
Jan 24, 2025



In image enhancement tasks, such as low-light and underwater image enhancement, a degraded image can correspond to multiple plausible target images due to dynamic photography conditions, such as variations in illumination. This naturally results in a one-to-many mapping challenge. To address this, we propose a Bayesian Enhancement Model (BEM) that incorporates Bayesian Neural Networks (BNNs) to capture data uncertainty and produce diverse outputs. To achieve real-time inference, we introduce a two-stage approach: Stage I employs a BNN to model the one-to-many mappings in the low-dimensional space, while Stage II refines fine-grained image details using a Deterministic Neural Network (DNN). To accelerate BNN training and convergence, we introduce a dynamic \emph{Momentum Prior}. Extensive experiments on multiple low-light and underwater image enhancement benchmarks demonstrate the superiority of our method over deterministic models.
Optimally-Weighted Maximum Mean Discrepancy Framework for Continual Learning
Jan 21, 2025Continual learning has emerged as a pivotal area of research, primarily due to its advantageous characteristic that allows models to persistently acquire and retain information. However, catastrophic forgetting can severely impair model performance. In this study, we tackle the issue of network forgetting by introducing a novel framework termed Optimally-Weighted Maximum Mean Discrepancy (OWMMD), which imposes penalties on representation alterations via a Multi-Level Feature Matching Mechanism (MLFMM). Furthermore, we propose an Adaptive Regularization Optimization (ARO) strategy to refine the adaptive weight vectors, which autonomously assess the significance of each feature layer throughout the optimization process. We conduct a comprehensive series of experiments, benchmarking our proposed method against several established baselines. The empirical findings indicate that our approach achieves state-of-the-art performance.
Incrementally Learning Multiple Diverse Data Domains via Multi-Source Dynamic Expansion Model
Jan 15, 2025



Continual Learning seeks to develop a model capable of incrementally assimilating new information while retaining prior knowledge. However, current research predominantly addresses a straightforward learning context, wherein all data samples originate from a singular data domain. This paper shifts focus to a more complex and realistic learning environment, characterized by data samples sourced from multiple distinct domains. We tackle this intricate learning challenge by introducing a novel methodology, termed the Multi-Source Dynamic Expansion Model (MSDEM), which leverages various pre-trained models as backbones and progressively establishes new experts based on them to adapt to emerging tasks. Additionally, we propose an innovative dynamic expandable attention mechanism designed to selectively harness knowledge from multiple backbones, thereby accelerating the new task learning. Moreover, we introduce a dynamic graph weight router that strategically reuses all previously acquired parameters and representations for new task learning, maximizing the positive knowledge transfer effect, which further improves generalization performance. We conduct a comprehensive series of experiments, and the empirical findings indicate that our proposed approach achieves state-of-the-art performance.
Information-Theoretic Dual Memory System for Continual Learning
Jan 13, 2025



Continuously acquiring new knowledge from a dynamic environment is a fundamental capability for animals, facilitating their survival and ability to address various challenges. This capability is referred to as continual learning, which focuses on the ability to learn a sequence of tasks without the detriment of previous knowledge. A prevalent strategy to tackle continual learning involves selecting and storing numerous essential data samples from prior tasks within a fixed-size memory buffer. However, the majority of current memory-based techniques typically utilize a single memory buffer, which poses challenges in concurrently managing newly acquired and previously learned samples. Drawing inspiration from the Complementary Learning Systems (CLS) theory, which defines rapid and gradual learning mechanisms for processing information, we propose an innovative dual memory system called the Information-Theoretic Dual Memory System (ITDMS). This system comprises a fast memory buffer designed to retain temporary and novel samples, alongside a slow memory buffer dedicated to preserving critical and informative samples. The fast memory buffer is optimized employing an efficient reservoir sampling process. Furthermore, we introduce a novel information-theoretic memory optimization strategy that selectively identifies and retains diverse and informative data samples for the slow memory buffer. Additionally, we propose a novel balanced sample selection procedure that automatically identifies and eliminates redundant memorized samples, thus freeing up memory capacity for new data acquisitions, which can deal with a growing array of tasks. Our methodology is rigorously assessed through a series of continual learning experiments, with empirical results underscoring the effectiveness of the proposed system.
ProteinWeaver: A Divide-and-Assembly Approach for Protein Backbone Design
Nov 08, 2024Nature creates diverse proteins through a `divide and assembly' strategy. Inspired by this idea, we introduce ProteinWeaver, a two-stage framework for protein backbone design. Our method first generates individual protein domains and then employs an SE(3) diffusion model to flexibly assemble these domains. A key challenge lies in the assembling step, given the complex and rugged nature of the inter-domain interaction landscape. To address this challenge, we employ preference alignment to discern complex relationships between structure and interaction landscapes through comparative analysis of generated samples. Comprehensive experiments demonstrate that ProteinWeaver: (1) generates high-quality, novel protein backbones through versatile domain assembly; (2) outperforms RFdiffusion, the current state-of-the-art in backbone design, by 13\% and 39\% for long-chain proteins; (3) shows the potential for cooperative function design through illustrative case studies. To sum up, by introducing a `divide-and-assembly' paradigm, ProteinWeaver advances protein engineering and opens new avenues for functional protein design.
DPLM-2: A Multimodal Diffusion Protein Language Model
Oct 17, 2024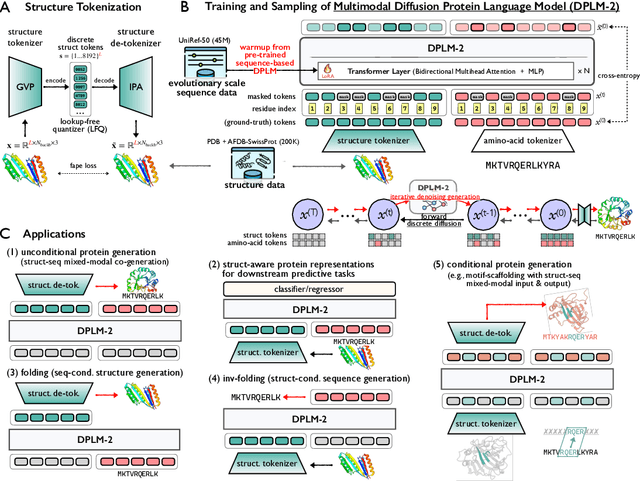
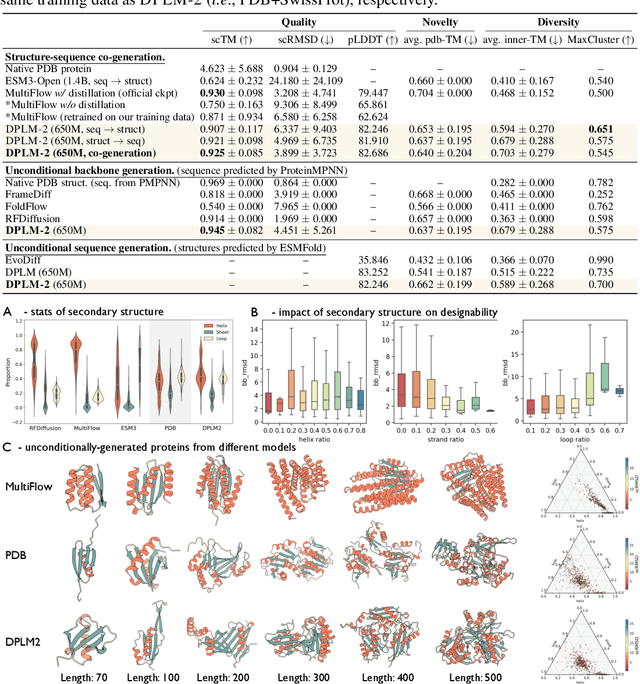
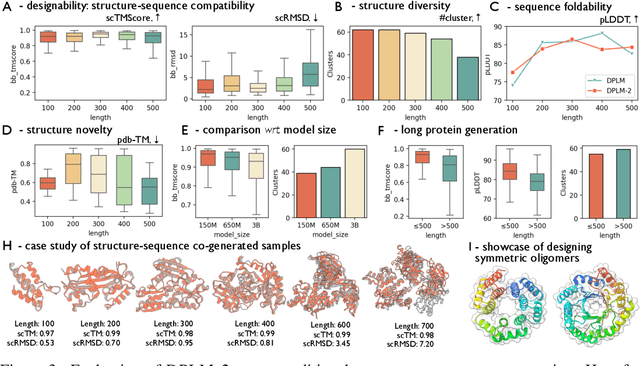
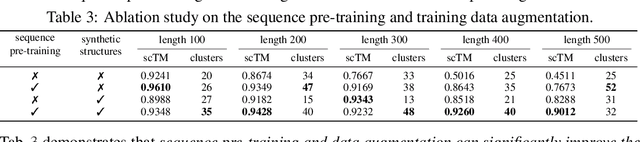
Proteins are essential macromolecules defined by their amino acid sequences, which determine their three-dimensional structures and, consequently, their functions in all living organisms. Therefore, generative protein modeling necessitates a multimodal approach to simultaneously model, understand, and generate both sequences and structures. However, existing methods typically use separate models for each modality, limiting their ability to capture the intricate relationships between sequence and structure. This results in suboptimal performance in tasks that requires joint understanding and generation of both modalities. In this paper, we introduce DPLM-2, a multimodal protein foundation model that extends discrete diffusion protein language model (DPLM) to accommodate both sequences and structures. To enable structural learning with the language model, 3D coordinates are converted to discrete tokens using a lookup-free quantization-based tokenizer. By training on both experimental and high-quality synthetic structures, DPLM-2 learns the joint distribution of sequence and structure, as well as their marginals and conditionals. We also implement an efficient warm-up strategy to exploit the connection between large-scale evolutionary data and structural inductive biases from pre-trained sequence-based protein language models. Empirical evaluation shows that DPLM-2 can simultaneously generate highly compatible amino acid sequences and their corresponding 3D structures eliminating the need for a two-stage generation approach. Moreover, DPLM-2 demonstrates competitive performance in various conditional generation tasks, including folding, inverse folding, and scaffolding with multimodal motif inputs, as well as providing structure-aware representations for predictive tasks.
ProteinBench: A Holistic Evaluation of Protein Foundation Models
Sep 10, 2024Recent years have witnessed a surge in the development of protein foundation models, significantly improving performance in protein prediction and generative tasks ranging from 3D structure prediction and protein design to conformational dynamics. However, the capabilities and limitations associated with these models remain poorly understood due to the absence of a unified evaluation framework. To fill this gap, we introduce ProteinBench, a holistic evaluation framework designed to enhance the transparency of protein foundation models. Our approach consists of three key components: (i) A taxonomic classification of tasks that broadly encompass the main challenges in the protein domain, based on the relationships between different protein modalities; (ii) A multi-metric evaluation approach that assesses performance across four key dimensions: quality, novelty, diversity, and robustness; and (iii) In-depth analyses from various user objectives, providing a holistic view of model performance. Our comprehensive evaluation of protein foundation models reveals several key findings that shed light on their current capabilities and limitations. To promote transparency and facilitate further research, we release the evaluation dataset, code, and a public leaderboard publicly for further analysis and a general modular toolkit. We intend for ProteinBench to be a living benchmark for establishing a standardized, in-depth evaluation framework for protein foundation models, driving their development and application while fostering collaboration within the field.
Dynamic Multi-Objective Lion Swarm Optimization with Multi-strategy Fusion: An application in 6R robot trajectory
May 31, 2024The advancement of industrialization has fostered innovative swarm intelligence algorithms, with Lion Swarm Optimization (LSO) being notable for its robustness and efficiency. However, multi-objective variants of LSO struggle with poor initialization, local optima entrapment, and slow adaptation to dynamic environments. This study proposes a Dynamic Multi-Objective Lion Swarm Optimization with Multi-strategy Fusion (MF-DMOLSO) to overcome these challenges. MF-DMOLSO includes an initialization unit using chaotic mapping, a position update unit enhancing behavior patterns based on non-domination and diversity, and an external archive update unit. Evaluations on benchmark functions showed MF-DMOLSO outperformed existing algorithms achieving an accuracy that exceeds the comparison algorithm by 90%. Applied to 6R robot trajectory planning, MF-DMOLSO optimized running time and maximum acceleration to 8.3s and 0.3pi rad/s^2, respectively, achieving a set coverage rate of 70.97% compared to 2% by multi-objective particle swarm optimization, thus improving efficiency and reducing mechanical dither.